Česká a slovenská psychiatrie

Časopis
Psychiatrické společnosti ČLS JEP
a Psychiatrickej spoločnosti SLS
souborný článek / review article
- AKTUÁLNÍ ČÍSLO
- ARCHIV
- VYHLEDÁVÁNÍ
- PERIODIKUM
- REDAKČNÍ RADA
- PŘEDPLATNÉ
- INZERCE
- AKTUALIZOVANÉ
POKYNY
PRO AUTORY
- ISSN 1212-0383




- © Česká a Slovenská psychiatrie 2025
- © Galén 2025
ÚLOHA MITOCHONDRIÍ V MECHANISMECH SYNAPTICKÉ PLASTICITY, BUNĚČNÉHO POŠKOZENÍ A PORUCH NÁLADY
ROLE OF MITOCHONDRIA IN MECHANISMS OF SYNAPTIC PLASTICITY, CELL DAMAGE AND MOOD DISORDERS
Zdeněk Fišar, Jana Hroudová, Jiří Raboch
Psychiatrická klinika 1. LF UK a VFN, Praha
SOUHRN
Fišar Z, Hroudová J, Raboch J. Úloha mitochondrií v mechanismech synaptické plasticity, buněčného poškození a poruch nálady
Ačkoli dysfunkce monoaminergních neurotransmiterových systémů mají významnou úlohu v patofyziologii poruch nálady, jedná se zřejmě o následky jiných, primárnějších abnormalit v transdukci signálu. Nové teorie o patofyziologii deprese a mechanismech účinků antidepresiv předpokládají, že určující úlohu ve vyšších mozkových funkcích narušených při poruchách nálady mají změny v nitrobuněčných signálních cestách projevující se narušením neuroplasticity Primárním regulátorem těchto procesů mohou být mitochondrie. Předpokládá se proto, že mitochondriální dysfunkce jsou zahrnuty v patofyziologii poruch nálady. U řady neuropsychiatrických onemocnění, včetně poruch nálady, bylo pozorováno narušení aktivity mitochondriálních enzymů, účinků nitrobuněčného kalcia a energetického metabolismu neuronů, poškození mitochondriální DNA a ovlivnění mitochondriálních funkcí psychofarmaky. Mitochondriální hypotéza bipolární afektivní poruchy koresponduje s hypotézou neurotrofní a hypotézou neuroplasticity, a to především vzhledem k významné úloze mitochondrií v buněčné energetice, regulaci kalciových signálních cest, produkci kyslíkových radikálů a apoptóze, tedy v procesech určujících synaptickou plasticitu, poškození, obnovu, přežití či smrt neuronů.
Klíčová slova: depresivní porucha, neuroplasticita, excitotoxicita, oxidační stres, mitochondrie
SUMMARY
Fišar Z, Hroudová J, Raboch J. Role of mitochondria in mechanisms of synaptic plasticity, cell damage and mood disorders
While dysfunctions within monoaminergic neurotransmitter systems are likely to play an important role in pathophysiology of mood disorders, it probably represents the downstream effects of more primary abnormalities in signal transduction. New theories about the pathophysiology of depression and mechanisms of action of antidepressants proposes that regulation of intracellular signalling pathways manifested by disturbed neuroplasticity plays a critical role in higher-order brain functions. Mitochondria maybe primary regulators of these processes. It is supposed that mitochondrial dysfunctions are included in pathophysiology of mood disorders. Disturbances in activity of mitochondrial enzymes, effects of intracellular calcium and energy metabolism, damage of mitochondrial DNA, and action of psychotropics on mitochondria were observed in many neuropsychiatric illnesses, mood disorders included. Mitochondrial hypothesis of bipolar affective disorder corresponds to neurotrophic and neuroplasticity hypotheses because of an important role of mitochondria in cell energetic, regulation of calcium signalling pathway, production of reactive oxygen species and apoptosis, i.e. in processes determining synaptic plasticity, damage, repairing, survival or death of neurons.
Key words: depressive disorder, neuronal plasticity, excitotoxicity, oxidative stress, mitochondria
ÚVOD
Předpokládá se, že symptomy psychiatrických onemocnění jsou spojeny s narušením přenosu, ukládání a zpracování informací v centrálním nervovém systému (CNS). Na narušení těchto základních funkcí mozku se podílejí abnormality jak neurovývojové, tak neurochemické. Vzhledem k časovému průběhu normálních i patologických změn nálady lze předpokládat, že náchylnost ke vzniku poruch nálady je podmíněna vývojovými odchylkami vedoucími k odlišným vlastnostem neuronových sítí v mozku, zatímco jednotlivé symptomy jsou určeny neurochemicky, tj. změnami v transmisi a transdukci nervových signálů v určitých oblastech mozku. Jedná se o procesy, které jsou velmi proměnlivé, vzájemně související a ovlivňující se.
Dynamické vlastnosti CNS umožňující reakci a adaptaci na vnější i vnitřní podněty jsou označovány jako neuroplasticita. Na základě pozorování překryvu mezi buněčnými procesy provázejícími depresivní poruchy, neuroplasticitu, chronický stres a dlouhodobé podávání antidepresiv a stabilizátorů nálady vznikly neurotrofní hypotézy deprese. Vzhledem ke složitosti a dynamice mezibuněčných a nitrobuněčných procesů nejsou molekulární mechanismy podmiňující změny neuroplasticity a přežívání neuronů za normálních ani za patologických podmínek dostatečně známy. Významnou úlohu v těchto procesech mají nepochybně mitochondrie, jakožto zdroje adenozin-5'-trifosfátu (ATP), producenti reaktivních forem kyslíku (ROS, "reactive oxygen species", též označované jako volné kyslíkové radikály), regulátory cytosolového kalcia a iniciátory vnitřní cesty aktivace programované buněčné smrti (apoptózy). V této práci jsou shrnuty poznatky o společných molekulárních mechanismech provázejících poruchy nálady, neuroplasticitu, poškození neuronů a funkce mitochondrií, tedy poznatky vedoucí k mitochondriálním hypotézám poruch nálady.
PORUCHY NÁLADY
Neurotrofní hypotézy
Nové hypotézy poruch nálady a mechanismů účinků antidepresiv předpokládají, že poruchy nálady jsou způsobeny strukturálními a funkčními změnami neuronů a určitých molekul v mezibuněčných i nitrobuněčných signálních cestách,1 přičemž antidepresiva působí proti těmto změnám. Strukturální a funkční abnormality v mozcích osob trpících depresivní poruchou mohou být spojeny s nízkými koncentracemi růstových faktorů, jako je mozkový neurotrofní faktor (BDNE "brain-derived neurotrophic factor"), se zvýšenou aktivitou osy hypotalamus - hypofýza - kůra nadledvin (HPA, "hypothalamic-pituitary-adrenal") a s glutamátergní neurotoxicitou.2,3
Neurotrofní hypotéza deprese4 a její novější varianty, jako je hypotéza neuroplasticity,5 předpokládají, že určujícím patofyziologickým rysem deprese jsou narušené mechanismy neuroplasticity v určitých oblastech mozku, přičemž chronický stres a jiné podněty vyvolávající buněčný stres jsou významnými kauzálními faktory ve vývoji tohoto poškození. Dlouhodobé podávání antidepresiv a stabilizátorů nálady vede k opravě narušených mechanismů neuroplasticity; na molekulární úrovni dochází ke zvýšení aktivity transkripčního faktoru aktivovaného v odezvě na zvýšení koncentrace cyklického adenozinmonofosfátu (CREB protein, "cAMP response element binding protein"), větší expresi neurotrofního faktoru BDNF a jeho receptoru trkB ("tropomyosin-related kinase B") a následně ke zvýšení neuroplasticity a obnově buněčných funkcí.
S touto hypotézou koresponduje i zánětlivá a neurodegenerativní hypotéza,6 podle níž je zvýšená neurodegenerace a narušená neurogeneze při depresi způsobena zánětlivými procesy vztaženými k oxidačnímu a nitrozačnímu stresu, katabolitům tryptofanu v indolamin-2,3-dioxygenázové cestě, prozánětlivým cytokinům a sníženým ω-3 polynenasyceným kyselinám. Protizánětlivé účinky antidepresiv vedou k obnově neurogeneze, která může být snížena zánětlivými procesy.
Síťová hypotéza7 předpokládá, že poruchy nálady odrážejí problémy se zpracováním informací určitými neuronovými sítěmi v mozku; podávání antidepresiv nebo jiná léčba zmírňující depresivní symptomy účinkují přes postupné zlepšování zpracování informací v těchto sítích. Podle této hypotézy nejsou depresivní symptomy nebo antidepresivní účinky přímo vyvolány změnami koncentrací neurotrofinů a jiných signálních molekul v mozku, ale neurotrofiny mohou působit jako rozhodující prostředek v procesu adaptace neuronových sítí na podněty z vnějšího prostředí.8 Síťová hypotéza tedy vychází z neurotrofní hypotézy, ale zvýrazňuje význam zpracování vnějších informací v procesu obnovy mozkových funkcí v průběhu léčby poruch nálady.
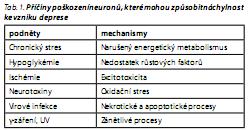
Výchozím předpokladem neurotrofní hypotézy a hypotéz na ni navazujících je tedy předpoklad, že narušení transdukce signálu v mozku vede k takovým změnám ve zpracování a uchovávání informací, které se projeví symptomy onemocnění. Příčiny poškození neuronů, které mohou způsobit náchylnost ke vzniku deprese, jsou shrnuty v tab. 1. Na řadě těchto mechanismů, jako je narušený energetický metabolismus, excitotoxicita, oxidační stres, nekróza a apoptóza, se významně podílejí mitochondrie.9
Poznatky podporující neurotrofní hypotézy pocházejí jednak z měření změn aktivity transkripčního faktoru CREB, neurotrofinu BDNF a dalších růstových faktorů při depresivní epizodě, jednak z pozorování mechanismů účinků dlouhodobého podávání antidepresiv a stabilizátorů nálady na aktivitu nitrobuněčných signálních cest vedoucí ke zvýšené produkci a aktivitě CREB a BDNF.10
Antidepresiva ovlivňují učení a paměť a zvyšují neuro-plasticitu a hipokampální neurogenezi.11,12 Regulují aktivitu signálních cest vztažených k neuroplasticitě přes zvýšení aktivity kaskády cyklický adenozinmonofosfát/proteinkináza A/CREB (cAMP/PKA/CREB), regulací aktivity proteinkináz závislých na kalciu a kalmodulinu typu II (PKCaMII) a upregulací kaskády proteinkináz aktivovaných mitogenem (MAPK).13 Hypotéza, že dlouhodobé podávání antidepresiv zvyšuje neuroplasticitu, je založena na upregulaci exprese růstových faktorů, především BDNF, v hipokampu a prefrontální kůře.10 Dosud ale chybějí velké longitudinální studie prokazující schopnost antidepresiv zvrátit atrofii mozkových struktur při depresi a bránit jí.
Pro stabilizátory nálady byla nalezena řada molekulárních cílů, především enzymy přímo či nepřímo inhibované lithiem (např. inozitolmonofosfatáza, glykogensyntázakináza-3 [GSK-3]) nebo valproátem (např. sukcinátsemialdehyddehydrogenáza, sukcinátsemialdehydreduktáza, histondeacyláza), cíle karbamazepinu (např. sodíkové kanály, adenozinové receptory, adenylátcykláza) a složky signálních cest regulované vícero léčivy.14 Neuroprotektivní účinky lithia a valproátu jsou založeny na ochraně proti glutamátergní neurotoxicitě, aktivaci cest podporujících přežití neuronů (např. cesty fosfoinozitid-3-kináza/proteinkináza B, PI3K/Akt) a indukci neurotrofních a neuroprotektivních proteinů (např. BDNF, Bcl-2, proteiny tepelného šoku).15
Polymorfismus genu pro BDNF byl asociován s depresí a bipolární afektivní poruchou. Byly pozorovány snížené koncentrace BDNF post mortem v mozcích depresivních osob. Rovněž koncentrace BDNF v krvi depresivních pacientů jsou zřejmě sníženy a po antidepresivní léčbě zvýšeny.8,16 Chronický stres, jakožto faktor podílející se často na vzniku depresivní poruchy, způsobuje přetrvávající supresi transkripce BDNF metylací histonů, zatímco antidepresiva obnovují syntézu BDNF acetylací histonů.17 Ve zvířecích modelech deprese bylo zjištěno, že lokální infúze BDNF do specifických oblastí mozku napodobuje antidepresivní účinky. Při depresi dochází k regulaci i jiných růstových faktorů.10
Avšak BDNF nevykazuje stejné účinky v celém mozku. Lokální infúze BDNF do určitých oblastí mozku vykazovala účinky podobné antidepresivním, ale infúzi do jiných oblastí (např. do ventral tegmental area, VTA) lze vztáhnout k indukci deprese. Úloha BDNF v cestě VTA - nucleus accumbens (NAc) je tedy zřejmě opačná než v hipokampu.18,19 Navíc snížená aktivita BDNF v obsáhlých oblastech předního mozku neindukovala sama o sobě chování podobné depresivnímu.10
Transkripční faktor CREB má v mozku účinky žádoucí i nežádoucí, v závislosti na oblasti mozku, kde působí. Aktivita CREB je zahrnuta v mechanismech učení a paměti a bylo potvrzeno, že její zvýšení v hipokampu koreluje se zvýšením exprese růstových faktorů, jako je BDNF, a způsobuje podobné účinky jako dlouhodobé podávání antidepresiv. Oproti tomu zvýšená aktivita CREB v NAc produkuje symptomy podobné depresivním. CREB a BDNF tedy obecně regulují plasticitu, proces, který není nezbytně dobrý či špatný, což je vidět v případu deprese.20 Strategie celkového farmakoterapeutického zvýšení aktivity CREB a BDNF v mozku se tedy nejeví jako optimální v léčbě neuropsychiatrických onemocnění.
Poškození mozkových struktur při depresi
Hlavní mozkové struktury spojované s neurobiologií deprese jsou součástí prefrontální kůry a limbického systému: kůra orbitofrontální, dorsolaterální a předního cingula, amygdala a hipokampus. V posledních desetiletích byly pomocí metod založených na magnetické rezonanci získány nové důkazy o poškození mozkových struktur při různých psychiatrických onemocněních; tato poškození zřejmě souvisejí s narušením procesů neuronální plasticity.
Kognitivní poškození při těžké depresi zahrnuje jednak narušenou koncentraci a pozornost, jednak deficit v explicitní paměti (tj. ve vědomém a záměrném vybavování si dřívějších zkušeností a informací). Tyto poruchy lze dát do souvislosti s abnormalitami funkce dorsolaterální prefrontální kůry, hipokampu a mediálního temporálního laloku. Atrofie hipokampu byla pozorována při rekurentní a těžké depresi21 i při posttraumatické stresové poruše.22 Měřením post mortem bylo zjištěno, že se nemění celkový počet neuronů a glií v hipokampu, ale je redukována velikost neuronů a objem neuropilu.23 Meta-analýzou dat o objemu hipokampu u pacientů s těžkou depresivní poruchou získaných zobrazením pomocí magnetické rezonance (MRI) bylo zjištěno, že ke změnám v objemu hipokampu dochází jen u osob s opakovanými epizodami deprese, kdy onemocnění trvá déle než dva roky, přičemž se tento efekt projevil u osob v dětském, středním a starším věku, ale nikoli u mladých dospělých osob.24 Znamená to, že atrofie hipokampu není znak náchylnosti k onemocnění ("trait marker"). Pro potvrzení tohoto závěru ale zatím chybí longitudinální studie.
Při bipolární afektivní poruše byla pomocí MRI a voxel-based morfometrie zjištěna redukce šedé hmoty v kůře předního cingula a bilaterální insuly. 25 Jedná se o změny v paralimbických oblastech zahrnutých v regulaci emocí.
Na modulaci uvolňování monoaminů a kortikosteroidů v odezvě na různé podněty se podílí amygdala. Meta-analýza výsledků měření změn objemu amygdaly při poruchách nálady ukázala, že objemy amygdaly u osob s bipolární či unipolární afektivní poruchou a u kontrolních osob nejsou výrazně odlišné.26 Objemy amygdaly tedy zřejmě nejsou spojeny s poruchami nálady.
Vzhledem k výskytu anhedonie, snížené motivace a snížené energetické hladiny při depresi se v její patofyziologii a symptomatologii předpokládá účast mesolimbického dopaminového systému. Dalšími mozkovými strukturami, jejichž funkce, struktura a plasticita může být narušena, jsou proto NAc a jeho dopaminergní vstup zVTA.19
Celkově lze říci, že neuropatologické a neurodegenerativní změny v mozku depresivních osob nejsou výrazné. Opakovaně byl zjištěn snížený počet gliových buněk ve frontálních korových oblastech osob s těžkou depresí nebo bipolární afektivní poruchou, což může být výsledek narušené gliogeneze. Byla navržena hypotéza, podle níž při progresi rekurentní depresivní poruchy vede kombinace genetických a vnějších faktorů nejprve k časné patologii gliových buněk (způsobené nadbytkem glukokortikoidů nebo nedostatkem neurotrofních a angiogenních faktorů) a poté k patologii neuronů (způsobené narušenou myelinizací a nadbytkem mimobuněčného glutamátu v důsledku jeho sníženého vychytávání gliemi).27 Snížená hustota neuronů je při depresi méně zřejmá než patologie gliových buněk, navíc je specifická pro určité frontální oblasti a je možné, že souvisí s odlišnou etiologií a patologií depresivních poruch u starších a mladších pacientů.28,29
NEUROPLASTICITA
Neuroplasticita popisuje funkční a strukturální změny neuronů a gliových buněk, které nastávají ve vyvíjejícím se i v dospělém mozku za účelem přizpůsobení se organismu vnějším i vnitřním podnětům.30,31 Jedná se o základní mechanismus adaptace neuronů. Neuroplasticita v dospělém mozku zahrnuje změny dendritických funkcí, reorganizaci synapsí, dlouhodobou potenciaci, dlouhodobou depresi, růst, větvení a rašení dendritů a axonu, synaptogenezi a neurogenezi.32 K neurogenezi dochází v dospělém mozku především v hipokampu, ale byla zjištěna i v dalších oblastech mozku, jako bulbus olfactorius a mozeček.33 Významnou úlohu v synaptické plasticitě a síle synapsí má hustota, velikost a tvar dendritických trnů. 34 Synaptické funkce zahrnují také obousměrnou komunikaci mezi neurony a astrocyty.27
Synaptickou plasticitou se rozumí především vývoj nových synapsí, změny v síle již existujících synapsí a eliminace synapsí. Synaptická plasticita je zřejmě buněčným základem učení a paměti.35 K dlouhodobé potenciaci (LTP) a dlouhodobé depresi (LTD) byly popsány další formy plasticity: vnitřní plasticita (určená např. fosforylací a distribucí iontových kanálů), inhibiční plasticita (daná funkcí inhibičních GABAergních neuronů) a homeostatická plasticita (zvyšující nebo snižující sílu všech synapsí na neuronu jako funkci aktivity).36 Další formou synaptické plasticity je metaplasticita (plasticita synaptické plasticity), která je indukována buněčnou aktivitou, ale projevuje se jako změna schopnosti indukovat následnou synaptickou plasticitu, jako je LTP nebo LTD; nemusí se tedy nutně projevovat změnou v účinnosti normální synaptické transmise. 37
Existuje jak postsynaptická, tak presynaptická plasticita. Výsledkem presynaptické plasticity je změna v uvolňování neurotransmiterů; 38 na jednom typu presynaptické plasticity se podílí i endokanabinoidní systém.39 Hlavní mechanismy postsynaptické plasticity typu LTP jsou spojeny s aktivací ionotropních glutamátových receptorů pro kyselinu (?)α-amino-3-hydroxy-5-metyl-4-izoxazol propionovou (AMPA) a N-metyl-D-aspartát (NMDA). AMPA receptory jsou odpovědné za depolarizaci membrány, která postačuje pro aktivaci NMDA receptorů, jejichž Ca2+-kanály jsou napěťově blokovány hořčíkem; poté dochází ke vtoku kalcia do buňky. 40 Kalcium spouští procesy vedoucí k synaptické plasticitě v dendritických trnech: 1. regulaci iontových kanálů, cytoskeletálních proteinů a syntézy nových proteinů; 2. změny vlastností synaptických AMPA receptorů; 3. reorganizaci aktinu a modulaci morfologie synaptických trnů; 4. iniciaci lokální syntézy proteinů v trnech a dendritech. Za vznik synaptické plasticity jsou tedy odpovědné glutamátové AMPA receptory, za její kontrolu NMDA receptory. Z hlediska doby trvání rozlišujeme krátkodobou a dlouhodobou synaptickou plasticitu. Krátkodobou synaptickou plasticitou se rozumějí změny v synaptické odezvě trvající desítky sekund až minuty. Klíčovým procesem LTP je vstup Ca2+ do buňky přes glutamátové ionotropní NMDA receptory (viz výše), aktivace proteinkináz PKCaMII, fosforylace AMPA receptorů v postsynaptické membráně, zabudování nitrobuněčně lokalizovaných AMPA receptorů do synaptické membrány a jejich laterální difúze do postsynaptické denzity (tj. aktivace spících synapsí, "unsilencing").13, 40 V odezvě na podnět vyvolávající LTD jsou AMPA i NMDA receptory odstraňovány ze synaptické membrány.
Dlouhodobá synaptická plasticita trvá hodiny až týdny, i déle. Vyžaduje indukci genové exprese a produkci a správné umístění nových molekul. Významnou úlohu zde má aktivace kaskády kináz regulovaných mimobuněčným signálem (ERK) a kaskády cAMP/PKA/CREB/BDNE13,20 Když jsou syntetizovány nové receptory, iontové kanály a jiné membránové molekuly, musí existovat mechanismus, jak je přemístit do specifického místa synaptické membrány - existuje tzv. synaptické značení ("tagging").41 Pro synaptické značení je určující aktivita cesty cAMP/PKA.
POŠKOZENÍ NEUROPLASTICITY
Oxidační stres
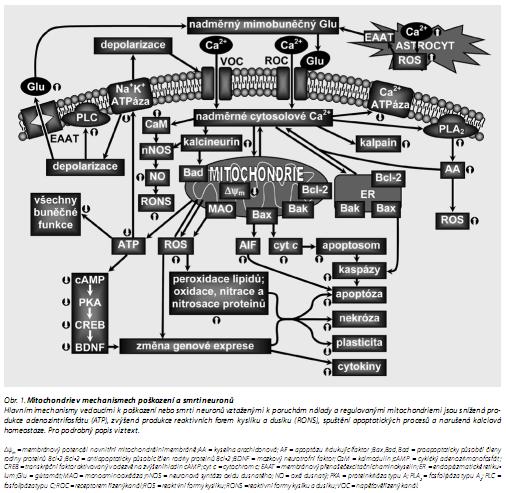
Volné radikály a jiné reaktivní látky mohou poškozovat membránu přes peroxidaci zbytků nenasycených mastných kyselin ve fosfolipidech tvořících lipidovou dvojnou vrstvu buněčné membrány. Dále mohou poškozovat další základní buněčné složky, což může vést k buněčné smrti nekrotickými či apoptotickými mechanismy.42,43 Oxidační poškození se podílí na společných komplexních interakcích mezi excitotoxicitou, apoptózou a zánětlivými procesy (obr. 1). Za oxidační stres jsou odpovědné hlavně reaktivní formy kyslíku a dusíku (RONS), jako je superoxid (O2 .-), peroxid vodíku (H2O2), hydroxyl (OH.), oxid dusnatý (NO.), peroxynitrit (ONOO-) a další.44 RONS jsou v nízkých koncentracích produkovány i za normálních fyziologických podmínek a aktivují různé buněčné signální cesty, které se účastní regulace přežití a smrti buňky43 Cíli redoxní signalizace jsou transkripční faktory, proteinkinázy a proteázy RONS mohou aktivovat transkripční faktory přímo (např. NF-κB, HSF1, p53), nebo přes fosforylaci regulací cesty PI3K/Akt a MAPK.45,46
Mozek je extrémně citlivý na oxidační poškození; jedním z důvodů je jeho vysoká spotřeba O2 (kolem 20 % celkové spotřeby). Kromě tvorby RONS a jiných reaktivních látek, jako je tomu ve všech buňkách, mohou v mozku vzniknout problémy s oxidačním stresem z těchto důvodů:44 1. zvýšené uvolňování excitačních aminokyselin v důsledku oxidačního stresu může spustit cyklus dalšího zvyšování produkce RONS; 2. zvýšená produkce superoxidu mitochondriemi v důsledku mutací a delecí mitochondriální DNA (mtDNA); 3. autooxidace některých neurotransmiterů; 4. uvolnění iontů železa a mědi při poškození mozku; 5. peroxidace neuronálních membránových lipidů (bohatých na zbytky polynenasycených mastných kyselin); 6. produkce H2O 2 monoaminooxidázami (MAO) ; 7. nepříliš velká antioxidační ochrana (nízké koncentrace katalázy); 8. produkce superoxidu, H2O 2 a cytokinů aktivovanými mikrogliemi; 9. zvýšení propustnosti hematoencefalické bariéry pro neurotoxiny reaktivními látkami; 10. neurotoxicita hemoglobinu atd.
Oxidační poškození DNA v neuronech je nejčastěji na úrovni bází a hlavní cestou pro opravu tohoto oxidačního poškození je vyštěpovací oprava báze (BER, "base excision repair"). Snížená funkce BER v mozkových buňkách by mohla být jednou z hlavních příčin normálního stárnutí, ale i řady neurologických abnormalit vedoucích např. k Alzheimerově nebo Parkinsonově nemoci.47
Ochranu mozku před účinky RONS zajišťují endogenní antioxidačními systémy, které zahrnují superoxiddismutázy (SOD, katalyzují redukci jednoho superoxidu na H2O2 a oxidaci druhého na O2), peroxiredoxiny a glutationperoxidázy (nejvýznamnější odstraňovače H2 O2), katalázy (v mozku nevýznamné odstraňovače H2O 2) a vychytávače reaktivních látek, jako je α-tokoferol a kyselina askorbová.44
Excitotoxicita
Vyčerpání energetických zásob (např. při ischémii, glutamátergní excitotoxicitě nebo chronickém stresu) vede ke snížené produkci ATP mitochondriemi, poškození procesů závislých na ATP a tím ke změněným buněčným funkcím (obr. 1). Nedostatečná funkce Na+K+-ATPáz vede k narušení iontových transmembránových gradientů, výtoku K+ a vtoku Na+, Cl- a Ca2+. Zvýšení mimobuněčných koncentrací K+ spouští depolarizaci membrán a obrácení funkce přenašečů pro aminokyseliny. Jsou aktivovány jak napěťově řízené (VOC), tak receptorem řízené (ROC) Ca2+-kanály a zvyšuje se koncentrace cytosolového Ca2+. Nitrobuněčné Ca2+ podporuje zvýšení mimobuněčných koncentrací excitačních aminokyselin, zvláště glutamátu, díky obrácení směru jeho přenosu membránovými přenašeči excitačních aminokyselin (EAAT, dříve známé jako glutamátové přenašeče); dochází tak k šíření neurotoxicity. Na zvýšení synaptických koncentrací glutamátu se může podílet i jeho uvolňování z astrocytů díky Ca2+-závislé exocytóze48 nebo inhibici EAAT (např. působením ROS) v gliových buňkách. Následně vazba glutamátu k NMDA a AMPA receptorům způsobuje nadměrný vtok Ca2+ do buňky, kalcium aktivuje fosfolipázy, proteázy a endonukleázy, které degradují membrány, proteiny a nukleové kyseliny nezbytné pro buněčnou integritu. Např. aktivace fosfolipázy A2 (PLA2) kalciem uvolňuje z membrán kyselinu arachidonovou (obr. 1), která potom přes cyklooxygenázovou a lipoxygenázovou cestu indukuje produkci superoxidu.49 Navíc vysoké nitrobuněčné koncentrace Ca2+ způsobují přetížení mitochondriálního Ca2+, zvýšenou produkci ROS a inhibici produkce ATP.5,42 Je možné, že určující úlohu v poškození mitochondrií, a tedy i celých buněk, v důsledku nadměrné akumulace kalcia mitochondriemi (např. při glutamátergní excitotoxicitě) má nedostatečná produkce ATP. Se vstupem iontů Na+ a Ca2+ přes chronicky aktivované NMDA a AMPA receptory je spojena aktivace Na+K+-ATPáz a Ca2+-ATPáz (obr. 1) a tyto procesy mohou využít celou energetickou kapacitu mitochondrií a tím zesílit excitotoxicitu.5
Excitotoxická nekróza zahrnuje nekontrolovaný vtok Na+ mající za následek rychlé nabobtnání a lýzu buňky. V poslední době bylo prokázáno, že glutamátem indukovaná smrt neuronů je spojena také s apoptózou, tj. charakteristickou fragmentací DNA, morfologickými změnami, aktivací kalpainu a kaspáz a upregulací nebo translokací apoptózu indukujícího faktoru (AIF) z mitochondrií do jádra. Glutamát ve vysokých koncentracích tedy může indukovat apoptózu jak mechanismy závislými na kaspázách, tak mechanismy nezávislými na těchto proteázách50,51 (obr. 1). Aktivované kaspázy-3 inaktivují také Ca2+-pumpu v plazmatické membráně, což vede k dalšímu přetížení nitrobuněčného Ca2+.52
Ochranu proti neurotoxicitě zajišťují neuronům sodíkové a kalciové pumpy (závislé na ATP), exprese proteinů vázajících Ca2+ a různé neurotrofní faktory (určující změny exprese NMDA receptorů, antioxidačních enzymů a antiapoptotických proteinů).50
Apoptóza
Programovaná buněčná smrt (PCD) má určující úlohu během vývoje i v normální buněčné homeostazi. Apoptóza je forma PCD popisovaná morfologickými změnami jako smrštění buňky, degradace cytoskeletu a následná tvorba váčkovitých výrůstků z cytoplazmatické membrány ("blebbing"), kondenzace chromatinu a fragmentace jádra a nakonec rozpad buňky na apoptotická tělíska. Při apoptóze také dochází k přesunu fosfatidylserinu z vnitřního na vnější povrch membrány, k poruchám propustnosti mitochondriální membrány a k uvolňování cytochromu c (cyt c).53 Dysregulace těchto procesů je zahrnuta v řadě onemocnění, včetně onemocnění neurodegenerativních. PCD je často užívána jako ekvivalent apoptózy, ale existují i neapoptotické formy PCD.54 PCD obecně označuje buněčnou smrt, která je zprostředkována nitrobuněčným programem, zatímco apoptóza (též označována jako PCD typu I nebo jako jaderná PCD) vykazuje navíc výše uvedené typické změny vzhledu buňky.
Za apoptózu jsou u savců odpovědné především kaspázy (cysteinové proteázy), které lze rozdělit na dva typy: iniciační (např. kaspázy-8, -9 a -10) a výkonné (např. kaspázy-3, -6 a -7).55 Hlavní kaspázy zahrnuté v apoptóze vedoucí ke smrti neuronů jsou kaspáza-9 a -3. Některé kaspázy mají zřejmě za fyziologických podmínek úlohu v neuronální plasticitě, jiné jsou aktivovány jen za patologických podmínek.
Klíčovou úlohu v regulaci nitrobuněčného apoptotického signálu mají proteiny z rodiny Bcl-2 ("B-cell lymphoma/leukemia-2 gene"), které zahrnují členy antiapoptotické (např. Bcl-2, Bcl-xL) i proapoptotické (např. Bax, Bak, Bad). Hlavní antiapoptotické faktory Bcl-2 a Bcl-xL jsou lokalizovány na vnější mitochondriální membráně, endoplazmatickém retikulu (ER) a perinukleární membráně. V CNS je Bcl-2 exprimován ve vysokých koncentracích během vývoje a je downregulován po narození. Oproti tomu Bcl-xL je také exprimován ve vyvíjejícím se mozku, ale jeho exprese se zvyšuje do dospělosti.56 Pro udržení přežívání neuronů jsou zřejmě určující Bcl-2, Bcl-xL a Bcl-w. Bcl-2 a Bcl-xL působí inhibicí proapoptotických členů Bcl-2 rodiny. Pro neuronální smrt je zřejmě určující aktivace Bax. Po indukci vnitřní cesty apoptózy je faktor Bax translokován do vnější mitochondriální membrány. Oligomerizací Bax a Bak (a možná dalších faktorů) jsou tvořeny mitochondriální apoptózu indukující kanály, které umožňují uvolňování cyt c a dalších proapoptotických faktorů (jako Smac/Diablo a AIF) s následnou aktivací kaspáz a buněčnou smrtí.57,58
Hlavní cesty zahrnuté do apoptózy lze rozdělit na vnější a vnitřní. 42 Vnější cesta začíná aktivací receptoru smrti (např. Fas) a vede k aktivaci kaspázy-8, která bud přímo aktivuje výkonné kaspázy (jako je kaspáza-3 a -7), nebo štěpí proapoptotický faktor Bid na tBid, který se přenáší do mitochondrií a indukuje vnitřní cestu apoptózy (aktivací Bax a Bak). Ve vnitřní cestě (obr. 1) způsobuje nadměrný vtok Ca2+ do nitrobuněčného prostoru změněnou funkci organel, jako jsou mitochondrie a ER. Aktivace Ca2+-závislých proteinfosfatáz (např. kalcineurinu) způsobuje translokaci proapoptotického faktoru Bad do mitochondrií a spuštění apoptózy sekvestrací antiapoptotických faktorů Bcl-2 a Bcl-xL. Uvolnění cyt c a dalších proapoptotických faktorů z mezimembránového prostoru mitochondrií vede k vytvoření apoptosomu vazbou cyt c k cytosolovému apoptotickému proteázu aktivujícímu proteinu (Apaf-1). Apoptosom štěpí prokaspázu-9 a kaspáza-9 potom aktivuje výkonné kaspázy.54,55 Kaspázy štěpí klíčové složky cytoskeletu a jádra, což vede k apoptotické smrti buňky. Za internukleosomální štěpení DNA je odpovědná kaspázou aktivovaná deoxyribonukleáza. Dále se z mitochondrií uvolňuje AIF a přenáší se do jádra a vede k apoptóze nezávislé na kaspázách.59
Významným zdrojem apoptotického signálu spojeného s aktivací kaspáz je ER.42, 54 ER je organela mající významnou úlohu v udržování nitrobuněčné Ca2+-homeostaze a ve správném skládání nově syntetizovaných proteinů. Signální mechanismy odpovídající za zapojení ER do apoptózy však nejsou dostatečně známy. Uvolňování Ca2+ z ER může také sekundárně aktivovat mitochondriální apoptózu.
Dalšími regulátory apoptózy jsou kalpainy (proteázy aktivované Ca2+ ), faktor p53, MAPK, rodina proteinů inhibujících apoptózu a proteiny tepelného šoku.42 Tumor supresorový a transkripční faktor p53 je hlavní modulátor odezvy na buněčný stress a jeho aktivace v odezvě na poškození DNA, oxidační stres, metabolické ohrožení nebo excitotoxicitu spouští apoptózu v mnoha typech buněk, včetně neuronů. Faktor p53 stimuluje expresi různých proapoptotických faktorů z rodiny Bcl-2, které po translokaci do mitochondrií způsobují uvolňování dalších proapoptotických faktorů s následnou aktivací kaspáz. Navíc může p53 podporovat buněčnou smrt přes transaktivaci receptoru smrti Fas nebo upregulaci Apaf-1. Kromě toho se může p53 přímo vázat na antiapoptotické faktory a inhibovat jejich funkci - tento proces se zřejmě uplatňuje při synaptické apoptóze.60
Chronický stres
Klinicky pozorovaný vztah mezi chronickým stresem a těžkou depresí lze vysvětlit jednak zvýšeným genetickým rizikem náchylných osob,61 jednak účinky stresu na neuroplasticitu a s ní související změny funkcí různých mozkových struktur. Nejdůkladněji je prozkoumán vliv stresu na synaptickou a morfologickou plasticitu hipokampu, neboť se jedná o strukturu mající významnou úlohu při učení a paměti a současně citlivou na stres díky vysoké koncentraci glukokortikoidních receptoru. Akutní a chronický stres mají zcela odlišné účinky na neuroplasticitu. Mírný stres po krátkou dobu zvyšuje poznávací schopnosti podporou synaptické plasticity v hipokampu, tj. LTP je optimální při mírně zvýšených koncentracích glukokortikoidů.62 Oproti tomu jsou při těžkém a dlouhotrvajícím stresu spuštěny procesy pro neuron škodlivé bud přímo, nebo přes zvýšenou neurotoxicitu jiných podnětů a poškození.63-66
Bylo potvrzeno, že chronický stres nebo dlouhodobé vystavení zvýšeným koncentracím glukokortikoidů poškozuje paměť závislou na hipokampu jak u experimentálních zvířat, tak u lidí.62,67 Dochází také k poškození hipokampu na úrovni morfologické neuroplasticity.68 Na škodlivých účincích glukokortikoidů se podílí inhibice uptake glukózy do neuronů, glutamátergní neurotoxicita a zvýšení koncentrací cytosolového kalcia a tvorby RONS. U hlodavců byla po vystavení stresu nebo glukokortikoidům pozorována i snížená hipokampální neurogeneze, která může mít úlohu v odezvě na podávání antidepresiv.69
Podobné účinky jako na hipokampus může mít chronický stres i na neuroplasticitu prefrontální kůry,70 zatímco účinky na amygdalu jsou spíše opačné.13 Chronický stres může způsobit v cestě VTA - NAc adaptivní změny, které mohou přispět k její dysregulaci při depresi. 19 Obecně mohou být účinky stresu na různé oblasti mozku velmi rozdílné.
Molekulární mechanismy indukované stresem a ovlivňující neuroplasticitu zahrnují především glutamátergní neurotoxicitu, změny v různých nitrobuněčných signálních cestách a sníženou expresi růstových faktorů. Při stresu dochází ke zvýšenému uvolňování glutamátu v hipokampu a prefrontální kůře,71,72 což vede k nadměrnému zvýšení cytosolového kalcia, nadměrné aktivitě enzymů závislých na Ca2+, degradaci cytoskeletu a buněčných proteinů a zvýšené tvorbě kyslíkových radikálů. Tyto procesy mohou způsobit atrofii nebo smrt neuronů.49,73 Stejnou cestu aktivuje také hypoxie-ischémie, epileptický záchvat či hypoglykémie. Neurony současně mobilizují řadu obranných mechanismů proti tomuto poškození.63
Akutní stres je spojen se zvýšenou mitochondriální biogenezí a enzymovou aktivitou při oxidační fosforylaci (OXPHOS). Oproti tomu při chronickém stresu dochází k poruchám v biogenezi, dysfunkci dýchacího řetězce, snížené produkci ATP, zvýšené produkci RONS, peroxidaci lipidů, poškození mitochondriální i jaderné DNA a zvýšené apoptóze či nekróze.74
V regulaci různých stresových systémů, jako je osa HPA, sympatický nervový systém, renin-angiotensinový systém a osa neuroendokrino-imunitní, může být zahrnut translokační protein (dříve označován jako periferní benzodiazepinový receptor), který je lokalizován ve vnější mitochondriální membráně, zvláště v kontaktních místech vnější a vnitřní mitochondriální membrány. V CNS je lokalizován hlavně v gliových buňkách a podílí se na regulaci biosyntézy steroidů a buněčných metabolických procesech. Tento translokační protein zřejmě participuje na některých neurodegenerativních onemocněních a na psychiatrických poruchách vztažených ke stresu, jako jsou úzkostné poruchy nebo posttraumatická stresová porucha.75
MITOCHONDRIE
Struktura a funkce mitochondrií
Mitochondrie jsou organely obsahující enzymy podílející se na oxidačním metabolismu eukaryontních buněk, tj. pyruvátdehydrogenázu, enzymy citrátového cyklu, enzymy katalyzující oxidaci mastných kyselin a enzymy a redoxní proteiny elektronového transportního řetězce a OXPHOS.
Mitochondrie jsou tvořeny vnější membránou, vnitřní membránou vytvářející kristy mezimembránovým prostorem a matrix.76 Vnější mitochondriální membrána obsahuje poriny proteiny vytvářející nespecifické póry, kterými volně difundují molekuly o hmotnosti do 5 kDa. Vnitřní mitochondriální membrána je propustná pouze pro kyslík, CO2 a vodu a umožňuje vytvoření iontových gradientů. V matrix se nacházejí enzymy oxidačního metabolismu, substráty, nukleotidové kofaktory, anorganické ionty, mitochondriální DNA (mtDNA) a RNA (mtRNA) a ribosomy.
Hlavní funkcí mitochondrií je produkce ATP pomocí enzymů citrátového cyklu a OXPHOS. Mitochondriální OXPHOS spotřebovává více než 80 % O2 a dodává kolem 92 % celkové buněčné energie. Dalšími důležitými funkcemi mitochondrií jsou produkce ROS, regulace nitrobuněčného kalcia, spuštění apoptózy, vývojová a synaptická plasticita a termogeneze.9,77
Oproti jiným typům buněk nevyužívají neurony glykolýzu k tvorbě ATP, pokud je mitochondriální bioenergetika nefunkční. Při inhibici mitochondriálního dýchání proto neurony rychle umírají, na rozdíl od astrocytů, které využívají i glykolyticky produkovaný ATP. Glukózový metabolismus v neuronech je směrován hlavně k pento-sofosfátovému cyklu, v němž vzniká redukovaný nikotin-amidadenindinukleotidfosfát (NADPH).78 NADPH je nezbytný k regeneraci glutationu a dalších složek obranného systému, který chrání buňku před oxidačním stresem přímou neutralizací ROS,79 nebo přes udržování exogenních antioxidantů, jako jsou vitaminy C a E, v aktivní formě. Antioxidanty mohou působit neuroprotektivně působením na komplexy I a III elektronového transportního řetězce, majícím za následek zvýšenou produkci ATP.
Mitochondrie a synaptická plasticita
Neurotransmise a synaptická plasticita jsou procesy energeticky velmi náročné, proto mají mitochondrie v jejich procesech podstatnou úlohu. Mitochondrie jsou velmi dynamické organely tj. jsou schopny přizpůsobovat svou odezvu podnětům z vnějšího či vnitřního prostředí a komunikovat mezi sebou a s jinými organelami. Mitochondrie jsou jednak aktivně transportovány do energeticky náročných částí neuronů, jako jsou synapse,80 jednak dochází k jejich rozdělování ("fission") a spojování ("fusion").81,82 Nerovnováha v dělení a fúzi mitochondrií může ovlivnit synapticou transmisi a plasticitu a mít vliv na přežití neuronu.83,84 Určující úlohu v dělení mitochondrií a neurodegeneraci mohou mít RONS, které inhibují mitochondriální dýchání.
Mitochondrie a oxidační stres
Mitochondrie jsou hlavním zdrojem buněčných ROS. Bylo identifikováno alespoň 10 míst schopných generovat superoxid; mezi nejvýznamnější patří komplexy I a III elektronového transportního řetězce.79,85 Rychlost tvorby ROS silně závisí na membránovém potenciálu a je inhibována při kyselém pH. 86 V izolovaných mitochondriích bylo zjištěno, že ke zvýšené tvorbě superoxidu v matrixu dochází, když mitochondrie nevytvářejí ATP a mají zvýšenou protonmotivní sílu (sestávající z membránového potenciálu a pH gradientu) a zvýšený poměr redukovaného a neredukovaného nikotinamidadenindinukleotidu (NADH/NAD+) nebo koenzymu Q (CoQH2/CoQ). 87 Biochemický mechanismus způsobující zvýšenou produkci ROS v odezvě na hypoxii není vnitřní vlastností mitochondrií, neboť v in vitro experimentech bylo pozorováno snížení generace ROS při velmi nízkých koncentracích O2. 88 V podmínkách in vitro 0,12-2 % kyslíku spotřebovaného mitochondriemi dávají vznik superoxidu. Produkce superoxidu mitochondriemi in vivo je však mnohem menší. Vzhledem k nedostatku poznatků o základních mitochondriálních funkcích in vivo nelze zatím určit ani množství, ani rychlost produkce superoxidu.87
Většina superoxidu je konvertována na H2O2 manganovou SOD nacházející se v mitochondriálním matrixu.79 H2O 2 vzniká také při metabolismu monoaminů mitochondriální MAO. Mitochondriální H2O2 difunduje do cytosolu a jádra a může být konvertován glutationperoxidázou a katalázou na vodu. Reakcí superoxidu s oxidem dusnatým vzniká peroxynitrit. Fentonovou reakcí (Fe 2+ + H2O2 -> Fe3+ + OH. + OH-) a rozkladem peroxynitritu vzniká vysoce reaktivní hydroxylový radikál (OH.), který může způsobit poškození neuronů a z toho plynoucí případné zdravotní problémy. 57,89,90 Mitochondrie obsahují i antioxidanty, jako koenzym Q10, kreatin a nikotinamid.
Poškození mitochondrií při narušeném energetickém metabolismu nebo při neurotoxicitě indukované glutamátem je způsobeno ROS, které produkují mitochondrie samotné (obr. 1). Lze rozlišovat ROS vyprodukované, uvolněné a vychytané obranným mitochondriálním systémem.79 Dochází k oxidačnímu poškození mitochondriálních proteinů, membrán a mtDNA, snížení syntézy ATP a tím k narušení řady metabolických funkcí. K významnému zvýšení ROS dochází jak při apoptóze, tak při nekróze.
Předpokládá se, že uvolnění cyt c z mezimembránového prostoru mitochondrií indukované proapoptotickým faktorem Bak vede k tvorbě ROS, které poté zvyšují propustnost vnější mitochondriální membrány (MOMP, "mitochondrial outer membrane permeabilization") a uvolňování cyt c, čímž se dále zvyšuje produkce ROS.91
ROS také indukují mitochondriální přechodné propustné póry (PTP, "mitochondrial permeability transition pores") měnící propustnost vnitřní mitochondriální membrány pro malé molekuly (do 1,5 kDa). Dochází ke ztrátě membránového potenciálu na vnitřní mitochondriální membráně (ΔΨm) vedoucí k MOMP a uvolňování proapoptotických faktorů. ROS mohou zvýšit propustnost vnější mitochondriální membrány také modifikací porinů typu aniontového kanálu závislého na napětí.
Mitochondrie v mozku jsou také důležitým cílem fyziologického a patofyziologického působení oxidu dusnatého (obr. 1); za jeho toxické působení je přitom odpovědný především peroxynitrit.92
Mitochondrie a kalcium
Cytoplazmatické kalcium je vychytáváno mitochondriemi a endoplazmatickým retikulem (ER) (obr. 1). Vnější mitochondriální membrána je propustná pro Ca2+, vnitřní membrána obsahuje přenašeče Ca2+: uniportér pro přenos Ca2+ do matrixu, Na+/Ca2+ a H+/Ca2+ antiportéry pro transport Ca2+ v opačném směru.5,77 Velký přísun Ca2+ do mitochondrií vede k depolarizaci jejich membrán, zastavení syntézy ATP, změně propustnosti mitochondriální membrány a uvolňování řady molekul včetně látek aktivujících apoptózu.93 Nabobtnání mitochondrií a ztráta elektrochemického potenciálu po vystavení oxidačnímu stresu nebo vysokým koncentracím Ca 2+ je důsledkem otevření megakanálů PTP ve vnitřní membráně.
Mitochondrie a buněčná smrt
Mitochondrie mají klíčovou úlohu v apoptotických procesech a tím i v patogenezi mnoha onemocnění, včetně neoplastických, neurodegenerativních a kardiovaskulárních. Vnitřní signální cesta apoptózy je spouštěna právě v mitochondriích (obr. 1). Změny při apoptóze zahrnují jednak zvýšení propustnosti vnější mitochondriální membrány pro cyt c, AIF a další faktory podílející se na indukci apoptózy, jednak ztrátu membránového potenciálu ΔΨm. Proteiny z rodiny Bcl-2 mají klíčovou úlohu v transdukci nitrobuněčného apoptotického signálu tím, že regulují permeabilitu vnější mitochondriální membrány. Mitochondriální apoptotické kaskády mohou být aktivovány lokálně v synapsích a dendritech, jedná se potom o tzv. synaptickou apoptózu.60
MITOCHONDRIÁLNÍ DYSFUNKCE A NEUROPSYCHIATRICKÁ ONEMOCNĚNÍ
Mitochondriální dysfunkce jsou způsobeny řadou komplexních mechanismů, zahrnujících jak genetické faktory z jaderného i mitochondriálního genomu, tak vliv vnějšího prostředí (západní styl života, stárnutí, léčiva a toxické látky).94 Výskyt mitochondriálních onemocnění v populaci není přesně znám, ale minimální prevalence se odhaduje na 1 : 5000, s tím, že může být mnohem vyšší.95 Specifické pro mitochondrie je, že při jejich dělení se kopie mtDNA rozdělí náhodně mezi dvě nové mitochondrie. Pokud je poškozených kopií mtDNA málo (heteroplazmická mutace), může se stát, že tyto kopie zůstanou jen v jedné z nových mitochondrií. K funkčním projevům mutace v mtDNA dojde proto pouze tehdy, když podíl poškozených molekul mtDNA dosáhne určité prahové hodnoty.96
Na základě studií zaměřených na komorbiditu poruch nálady a somatických symptomů byly nalezeny asociace s bipolární afektivní poruchou např. pro mutace mtDNA 3644T->C ovlivňující komplex I dýchacího řetězce,97 3243A->G ovlivňující mitochondriální tRNA98 nebo mutace jaderného genu pro mtDNA polymerázu.99 Pozornost byla věnována možnosti, že mitochondriální dysfunkce mohou ovlivnit kalciové signální cesty způsobem vedoucím k poruchám nálady.99,100
Syndrom mitochondriální encefalomyopatie, laktátová acidóza a iktu podobné příhody (MELAS, "mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes"), který je nejčastěji způsoben mutací 3243A->G mitochondriálního genomu, zahrnuje záchvaty, encefalopatii a mrtvici podobné epizody, ale také sekundární neuropsychiatrické projevy, jako kognitivní poškození, bolesti hlavy, deprese a další.101
Dosavadní zprávy o komorbiditě psychiatrických onemocnění a mitochondriálních poruch se týkají popisu relativně malých souborů osob, nebo dokonce jen jednotlivých případů. Většinou se jedná o demence, psychotická onemocnění nebo poruchy nálady, jejichž projevy obvykle předcházejí diagnózu mitochondriálního onemocnění.102,103 Např. ve skupině 35 dětí s biochemicky a geneticky potvrzenou mitochondriální poruchou se v 5 případech vyskytla těžká deprese předcházející diagnózu mitochondriální poruchy.104 Ve skupině 205 diabetiků jich bylo nalezeno 9, kteří měli mutaci 3243A->G. Z nich 4 měli i diagnózu duševní poruchy (těžká deprese, fobické úzkostné poruchy). U části pacientů se syndromem MELAS byl popsán výskyt depresivní poruchy nebo fobických úzkostných poruch105 nebo obsedantně-kompulzivní poruchy106 Analýza post mortem vzorků mozku rovněž naznačila, že akumulace mutací mtDNA 3243A->G by mohla mít úlohu v patofyziologii bipolární afektivní poruchy a schizofrenie.98 Předpokládá se, že mitochondriální dysfunkce jsou hlavním společným faktorem podmiňujícím asociaci migrény s těžkou depresivní poruchou, bipolární afektivní poruchou, panickou poruchou a sociální fobií,107 které se vyskytují s migrénou více než dvakrát častěji než bez ní.108 Pozornost je věnována také skutečnosti, že mitochondriální dysfunkce jsou kauzálně vztaženy k inzulínové rezistenci a metabolickému syndromu,94 a metabolický syndrom a jeho složky jsou asociovány s depresivní symptomatikou.109
Mitochondriální dysfunkce vedou k oxidačnímu stresu, poškození a delecím mtDNA, narušení kalciové homeostaze, změněné morfologii mitochondrií, změnám v dělení a spojování mitochondrií a případně až ke smrti neuronu. Mitochondrie tak mohou přispět k řadě neurodegenerativních a psychiatrických onemocnění. Podíl mitochondriálních dysfunkcí byl popsán v patogenezi Alzheimerovy, Parkinsonovy a Huntingtonovy nemoci, mrtvice, amyotropní laterální sklerózy, a také u schizofrenie, depresivní poruchy a bipolární afektivní poruchy.9,44,50, 57,77,103,110-113
Látky, které by mohly bránit poškození a dysfunkcím mitochondrií, a které jsou proto zahrnuty v terapeutických strategiích farmakologické léčby různých neurodegenerativních onemocnění, lze rozdělit na vitaminy a antioxidanty (thiamin, riboflavin, niacin, kyselina pantothenová, pyridoxin, biotin, folát, kyselina askorbová, tokoferoly, karotenoidy, CoQ10, kyselina lipoová, acetylcystein) a bio-energetické látky (karnitiny, kreatin, pyruvát, sirtuiny, nikotinamid a další). Pro vyšší účinnost v mitochondriích byly některé antioxidanty konjugovány s trifenylfosfoniem (především deriváty CoQ10). 57,114,115 Stopové prvky, jako železo, měď, zinek, mangan nebo selen, jsou nezbytné pro některé mitochondriální funkce a jejich nedostatek či nadbytek může být spojen s dysfunkcemi.116,117 Mnohé antioxidanty účinkují na zvířecích modelech, ale nikoli u lidí. Obecně nemusejí být vysoké koncentrace antioxidantů dobré a mohou způsobit i buněčné poškození. 118 Např. podávání (β-karotenu, vitaminu A, vitaminu C, vitaminu E a selenu bylo bez významných účinků na výskyt gastrointestinálních nádorů; naopak bylo pozorováno zvýšení celkové mortality.119 Existuje však i řada důkazů o tom, že dysfunkce mitochondriálních enzymů mohou být zlepšeny odpovídajícím příjmem substrátů a prekurzorů koenzymů, a tyto látky tedy mohou být užitečné v prevenci poškození mitochondrií při Alzheimerově či Parkinsonově nemoci.116,120
Předpoklad, že mitochondriální dysfunkce by mohly mít úlohu v patofyziologii bipolární afektivní poruchy, vychází z konceptu, že se jedná o geneticky ovlivněnou poruchu funkcí synapsí a neuronálních sítí, spíše než o pouhý nedostatek či nadbytek určitých neurotransmiterů.103,121 Naprostá většina bipolárních pacientů nevykazuje symptomy klasických mitochondriálních nemocí, tj. poruch způsobených poškozením v mitochondriálním dýchacím řetězci.96,122 Předpokládá se, že malé poruchy mitochondriálních funkcí mohou způsobovat změny synaptické plasticity a narušenou buněčnou odolnost vedoucí ke vzniku symptomů bipolární afektivní poruchy.9
Důkazy pro účast mitochondriálních dysfunkcí v patofyziologii psychiatrických poruch byly v poslední době shrnuty v několika přehledech.123-125 Pro bipolární afektivní poruchu tyto důkazy zahrnují:
- 1. narušení aktivity mitochondriálních enzymů,126,127
- 2. poškození kalciových signálních cest,9,99,100
- 3. poškození energetického metabolismu,128
- 4. zvýšení delecí, mutací nebo polymorfismů mtDNA v mozku,97,98,121
- 5. abnormální expresi klíčových mitochondriálních proteinů kódovaných jadernou DNA,129
- 6. účinky psychotropních látek na mitochondrie,9,111
- 7. komorbiditu poruch nálady u určitých mitochondriálních poruch.102,103, 130
Vliv antidepresiv a stabilizátorů nálady na mitochondriální funkce
Nejlépe prostudovaným účinkem různých antidepresiv na aktivitu mitochondriálních enzymů je inhibice MAO.131 Přímé či nepřímé účinky antidepresiv na další mitochondriální enzymy jsou známy poměrně málo. Vzhledem k tomu, že mitochondrie jsou známým cílem působení oxidu dusnatého, mohou tricyklická antidepresiva vykazovat své protizánětlivé a neurotrofní účinky přes sníženou produkci NO a prozánětlivých cytokinů.132 Byly pozorovány značně rozdílné účinky nefazodonu, trazodonu a buspironu na indukci mitochondriálních dysfunkcí a cytotoxicity133 Studium přímého vlivu řady farmakologicky odlišných antidepresiv (desipraminu, amitriptylinu, imipraminu, citalopramu, venlafaxinu, mirtazapinu, tianeptinu a moklobemidu) a stabilizátorů nálady (lithia, valproátu a olanzapinu) na aktivitu mitochondriálních enzymů in vitro ukázalo jejich výrazné inhibiční účinky na komplex I a komplex IV elektronového transportního řetězce.134
Augmentace vedoucí ke zvýšení folátu nebo snížení homocysteinu by mohla být užitečná při farmakoterapii antidepresivy, neboť nízký folát a zvýšený homo cystein zvyšují produkci ROS a přispívají tak k mitochondriálním dysfunkcím.135
Hlavní účinky stabilizátorů nálady na regulaci mitochondriálních funkcí in vivo se předpokládají nepřímé. Např. dlouhodobé podávání lithia zvyšuje koncentrace antiapoptotického proteinu Bcl-2, snižuje koncentrace proapoptotického proteinu p53 a inhibuje GSK-3.136 Inhibice GSK-3 potom může přes deinhibici komplexu mitochondriální pyruvátdehydrogenázy 137 zvyšovat maximální metabolickou rychlost v mozku. Lithium zřejmě mění aktivitu komplexů dýchacího řetězce in vivo i přímým působením.138,139 Předpokládá se proto, že neurotrofní a neuroprotektivní účinky lithia jsou uskutečňovány přes jeho účinky na mitochondriální funkce.9,140
Mitochondriální hypotézy poruch nálady
Na základě řady měření mozkových procesů souvisejících s mitochondriálními funkcemi a dysfunkcemi při poruchách nálady (tab. 2) byla navržena hypotéza, podle níž jsou jemné mitochondriální dysfunkce důležitou součástí bipolární afektivní poruchy a obnova či zvýšení těchto funkcí mohou být klíčové v léčbě onemocnění.9 Jinak řečeno, plasticita a energetika synapsí je při poruchách nálady pozměněna a modulátorem odpovědným za účinnost či neúčinnost různých antidepresiv jsou mitochondrie. Podklady pro formulaci této hypotézy pocházejí především ze studia účinků antidepresiv a stabilizátorů nálady na mitochondriální funkce a z in vivo měření změn buněčné energetiky v mozcích depresivních osob (pomocí magnetické rezonance molekul obsahujících fosfor).

Při bipolární afektivní poruše byly v mozku nalezeny změny v koncentracích N-acetylaspartátu, glutamátu a glutaminu, složek obsahujících cholin, myoinozitolu, laktátu, fosfokreatinu, fosfomonoesterů a nitrobuněčného pH. 141 Především na základě těchto in vivo měření byla formulována bioenergetická hypotéza mitochondriální dysfunkce při bipolární afektivní poruše113 předpokládající při tomto onemocnění narušení oxidační fosforylace, posun ke glykolytické produkci energie, sníženou produkci ATP, změněné koncentrace fosfomonoesterů a změněný metabolismus fosfolipidů (tab. 2).
Kalciová hypotéza mitochondriální dysfunkce při bipolární afektivní poruše je založena jednak na narušené kalciové homeostazi při neurodegenerativních onemocněních a poruchách nálady,142,143 jednak na pozorování mutací mtDNA v mozku, asociace polymorfismů mtDNA a bipolární poruchy a změn v expresi genů vztažených k mitochondriím v mozku.144 Podle této hypotézy polymorfismy nebo mutace mtDNA a delece mtRNA způsobene mutacemi jaderných genů mohou způsobit dysregulaci kalcia mitochondriemi, což může vést k symptomům onemocnění.103,128,144
ZÁVĚR
Důkazy o tom, že mitochondriální dysfunkce jsou zahrnuty v patofyziologii psychiatrických poruch, zahrnují narušenou aktivitu mitochondriálních enzymů, kalciových signálních cest a energetického metabolismu, zvýšené delece, mutace a polymorfismy mtDNA a účinky psychotropních látek na mitochondrie. Zdá se, že nedostatečné mitochondriální funkce jsou schopny spustit různá psychiatrická onemocnění, není však dosud jasné, zda mitochondriální dysfunkce přispívají k vývoji onemocnění, nebo zda se jedná o vedlejší jev.
Křížová propojení mezi nitrobuněčnými signálními cestami transdukce signálu ovlivněnými při poruchách nálady, účincích antidepresiv, stresu, apoptóze a synaptické plasticitě znamenají, že změny neuroplasticity mohou být vyvolány v různých místech různých signálních cest a že terapeutická strategie léčby poruch nálady by měla být optimálně zaměřena na více cílů a mechanismů. Předpokládá se, že významným terapeutickým cílem pro léčbu bipolární afektivní poruchy stabilizátory nálady by mohly být procesy oxidačního poškození kontrolované mitochondriemi.127
Unikátní vlastnosti mitochondrií je předurčují jako nitrobuněčné cíle nových léčiv selektivně modulujících OXPHOS, produkci ROS, propustnost mitochondriálních membrán, účinky Bcl-2 a expresi mtDNA.145 Farmakologická regulace mitochondriálních funkcí při poruchách nálady je teprve ve vývojovém stadiu, neboť není dostatek informací o vedlejších účincích takových látek. V současné době navíc neumíme farmakologicky ovlivnit pouze určitou oblast v mozku.
Použité zkratky: ΔΨm = membránový potenciál na vnitřní mitochondriální membráně; AA = kyselina arachidonová; AIF = apoptózu indukující faktor; Akt = proteinkináza B; AMPA = kyselina (± ) -α -amino-3-hydroxy-5-methyl-4-izoxazo propionová; Apaf-1 = apoptotický proteázu aktivující protein; ATP = adenozin-5'-trifosfát, adenozintrifosfát; Bax, Bad, Bad = proapoptoticky působící členy rodiny proteinů Bcl-2; Bcl-2 = rodina proteinů je zahrnuta v regulaci a poptotické buněčné smrti; Bcl-2, Bcl-xL, Bcl-w = antiapoptoticky působící členy rodiny proteinů Bcl-2; BDNF = mozkový neurotrofní faktor; BER = vyštěpovacíoprava báze; Bid/tBid = proapoptoticky faktor; CaM = kalmodulin; cAMP = cyklický adenozinmonofosfát; CNS = centrální nervový systém; CoQ = koenzym Q; CREB protein = transkripční faktor aktivovaný v odezvě na zvýšení hlad in cAMP; cyt c = cytochrom c; EAAT = membránový přenašeč excitačních aminokyselin; ER = endoplazmatické retikulum; ERK= kináza regulovaná mimobuněčným signálem; GABA= kyselina γ-aminomáselná; Glu = glutamát; GSK-3 = glykogensyntázakináza-3; HPA = hypotalamus-hypofýza-kůra nadledvin; LTD = dlouhodobá deprese; LTP = dlouhodobá potenciace; MAO = monoamino-oxidáza; MAPK= proteinkináza aktivovaná mitogenem; MELAS = mitochondriální encefalomyopatie, laktátová acidóza a iktu podobné příhody; MOMP = permeabilizace vnější mitochondriální membrány; MRI = zobrazení pomocí magnetické rezonance; mtDNA = mitochondriální DNA; mtRNA = mitochondriální RNA; NAc = nucleus accumbens; NAD = nikotinamidadenindinukleotid; NADPH = redukovaný nikotinamidadenindinukleotidfosfát; NF-κB = jaderný faktor-κB; NMDA = N-methyl-D-aspartát; nNOS = neuronová syntáza oxidu dusnatého; NO = oxid dusnatý; OXPHOS = oxidační fosforylace; PCD = programovaná buněčná smrt; PI3K = fosfoinozitid-3-kináza; PKA = proteinkináza typu A; PKCaM= protein kináza závislá na vápníku a kalmodulinu; PLA2 = fosfolipáza typu A2; PLC = fosfolipáza typu C; PTP = póry zvyšující propustnost mitochondriální membrány pro molekuly do molekulové hmotnosti 1500 daltonů; ROC = receptorem řízený kanál; ROS = reaktivní formy kyslíku; RONS = reaktivní formy kyslíku a dusíku; SOD = superoxiddismutáza; trkB = tropomyosin-related kinase B; VOC = napěťově řízený kanál; VTA = ventral tegmental area.
LITERATURA
- 1. Fišar Z, Hroudová J. Intracellular signalling pathways and mood disorders. Folia Biol 2010; 56 (4): 135-148.
- 2. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature 2008; 455 (7215): 894-902.
- 3. Mathew SJ, Manji HK, Charney DS. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology 2008; 33 (9): 2080-2092.
- 4. Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry 1997; 54 (7): 597-606.
- 5. Nicholls DG Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci 2008; 1147: 53-60.
- 6. Maes M, Yirmyia R, Noraberg J et al. The inflammatory & neuro degenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis 2009; 24 (1): 27-53.
- 7. Castrén E. Is mood chemistry? Nat Rev Neurosci 2005; 6 (3): 241-246.
- 8. Castrén E, Rantamáki T. Role of brain-derived neurotrophic factor in the aetiology of depression: implications for pharmacological treatment. CNS Drugs 2010; 24(1): 1-7. 9. Quiroz JA, Gray NA, Kato T, Manji HK. Mitochondrially mediated plasticity in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology 2008; 33 (11): 2551-2565.
- 10. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry 2006; 59 (12): 1116-1127.
- 11. Drzyzga LR, Marcinowska A, Obuchowicz E. Antiapoptotic and neurotrophic effects of antidepressants: a review of clinical and experimental studies. Brain Res Bull 2009; 79 (5): 248-257.
- 12. Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus 2006; 16 (3): 239-249.
- 13. Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 2008; 33 (1): 88-109.
- 14. Gould TD, Quiroz JA, Singh J, Zarate CA, Manji HK. Emerging experimental therapeutics for bipolar disorder: insights from the molecular and cellular actions of current mood stabilizers. Mol Psychiatry 2004; 9 (8): 734-755. 15 Chuang DM. The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci 2005; 1053: 195-204.
- 16. Castrén E, Voikar V, Rantamáki T. Role of neurotrophic factors in depression. Curr Opin Pharmacol 2007; 7 (1): 18-21.
- 17. Tsankova NM, Berton O, Renthal W et al. Sustained hippocampal chro-matin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 2006; 9: 519-525.
- 18. Eisch AJ, Bolaňos CA, de Wit J et al. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol Psychiatry 2003; 54(10): 994-1005.
- 19. Nestler EJ, Carlezon WA Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 2006; 59 (12): 1151-1159.
- 20. Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci 2005; 28 (8): 436-445.
- 21. Campbell S, Marriott M, Nahmias C, MacQueen GM. Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry 2004; 161 (4): 598-607.
- 22. Bremner JD, Elzinga B, Schmahl C, Vermetten E. Structural and functional plasticity of the human brain in posttraumatic stress disorder. Prog Brain Res 2008; 167: 171-186.
- 23. Stockmeier CA, Mahajan GJ, Konick LC et al. Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry 2004; 56 (9): 640-650.
- 24. McKinnon MC, Yucel K, Nazarov A, MacQueen GM. A meta-analysis examining clinical predictors of hippo-campal volume in patients with major depressive disorder. J Psychiatry Neurosci 2009; 34(1): 41-54.
- 25. Ellison-Wright I, Bullmore E. Anatomy of bipolar disorder and schizophrenia: a meta-analysis. Schizophr Res 2010; 117(1): 1-12.
- 26. Hajek T, Kopeček M, Kozeny J et al. Amygdala volumes in mood disorders - meta-analysis of magnetic resonance volumetry studies. J Affect Dis-ord2009; 115 (3): 395-410.
- 27. Rajkowska G, Miguel-Hidalgo JJ. Glio-genesis and glial pathology in depression. CNS Neurol Disord Drug Targets 2007; 6 (3): 219-233.
- 28. Rajkowska G, Miguel-Hidalgo JJ, Dubey P, Stockmeier CA, Krishnan KR. Prominent reduction in pyramidal neurons density in the orbitofrontal cortex of elderly depressed patients. Biol Psychiatry 2005; 58 (4): 297-306.
- 29. Van Otterloo E, O'Dwyer G, Stockmeier CA et al. Reductions in neuronal density in elderly depressed are region specific. Int J Geriatr Psychiatry 2009; 24 (8): 856-864.
- 30. Mesulam MM. Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron 1999; 24 (3): 521-529.
- 31. Nestler EJ, Barrot M, DiLeone RJ et al. Neurobiology of depression. Neuron 2002; 34(1): 13-25.
- 32. Fišar Z, Hroudová J. Common aspects of neuroplasticity, stress, mood disorders and mitochondrial functions. Activitas Nervosa Superior Rediviva 2010; 52(1): 3-20.
- 33. Ponti G, Peretto P, Bonfanti L. Genesis of neuronal and glial progenitors in the cerebellar cortex of peripuberal and adult rabbits. PLoS ONE. 2008; 3 (6): e2366. doi:10.1371/journal.pone.0002366. Dostupný z http://www.plosone.org/arti-cle/info%3Adoi%2F10.1371%2Fjournal.pone.0002366.
- 34. von Bohlen und Halbach O. Structure and function of dendritic spines within the hippocampus. Ann Anat 2009; 191 (6): 518-531.
- 35. Citri A, Malenka RC Synaptic plasticity: multiple forms, functions and mechanisms. Neuropsychopharmacology 2008; 33(1): 18-41.
- 36. Nelson SB, Turrigiano GG. Strength through diversity. Neuron 2008; 60 (3): 477-482.
- 37. Abraham WC, Bear ME Metaplastic-ity: the plasticity of synaptic plasticity. Trends Neurosci 1996; 19 (4): 126-130.
- 38. García-Junco-Clemente P, Linares-Clemente P, Fernández-Chacón R. Active zones for presynaptic plasticity in the brain. Mol Psychiatry 2005; 10 (2): 185-200.
- 39. Fišar Z. Phytocannabinoids and endocannabinoids. Curr Drug Abuse Rev. 2009; 2(1): 51-75.
- 40. Rebola N, Srikumar BN, Mulle C. Activity-dependent synaptic plasticity of NMDA receptors. J Physiol 2010; 588 (Pt 1): 93-99.
- 41. Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature 1997; 385 (6616): 533-536.
- 42. Nakka VP, Gusain A, Mehta SL, Raghubir R. Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol Neurobiol 2008; 37 (1): 7-38.
- 43. Novo E, Parola M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair 2008; 1 (1): 5. doi:10.1186/1755-1536-1-5. Dostupný z http://www.fibrogenesis.com/content/1/1/5.
- 44. Halliwell B. Oxidative stress and neuro degeneration: where are we now? J Neurochem 2006; 97 (6): 1634-1658.
- 45. Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 2001; 21(1): 2-14.
- 46. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biolo
Celá stať v dokumentu PDF
Čes a slov Psychiatr 2011;107(1): 14 -27