Česká a slovenská psychiatrie

Časopis
Psychiatrické společnosti ČLS JEP
a Psychiatrickej spoločnosti SLS
souborný článek / review article
- AKTUÁLNÍ ČÍSLO
- ARCHIV
- VYHLEDÁVÁNÍ
- PERIODIKUM
- REDAKČNÍ RADA
- PŘEDPLATNÉ
- INZERCE
- AKTUALIZOVANÉ
POKYNY
PRO AUTORY
- ISSN 1212-0383




- © Česká a Slovenská psychiatrie 2025
- © Galén 2025
ALZHEIMEROVA NEMOC: RIZIKOVÉ FAKTORY, BIOMARKERY A FARMAKOTERAPEUTICKÉ STRATEGIE
ALZHEIMER´S DISEASE: RISK FACTORS, BIOMARKERS, AND PHARMACOTHERAPEUTIC STRATEGIES
Zdeněk Fišar, Jana Hroudová
Psychiatrická klinika 1. LF UK a VFN v Praze
Práce byla podpořena grantem AZV MZ ČR NU23-04-00032.
SOUHRN
Fišar Z, Hroudová J. Alzheimerova nemoc: rizikové faktory, biomarkery a farmakoterapeutické strategie
Jsou shrnuty poznatky o rizikových faktorech a biomarkerech Alzheimerovy nemoci (AD), které jsou základem pro poznání patofyziologie onemocnění a pro rozpoznání buněčných cílů nových léčiv. Hlavními cíli nových léčiv pro AD jsou procesy spojené s neurotoxicitou amyloidu beta (A?) a tau proteinu, narušenou neurotransmisí, metabolickou dysregulací, mitochondriální dysfunkcí, oxidačním stresem, neurozánětem, neuroplasticitou, proteostází a proteinopatií. Některá nová léčiva jsou zaměřena na více asociovaných buněčných procesů, jako je cholinergní deplece, toxicita glutamátu, agregace A?, hyperfosforylace tau, oxidační stres a aktivita mitochondriálních proteinů. Farmakologická intervence při AD s potenciálem být kauzální zahrnuje regulaci produkce, eliminace, šíření a vzájemné interakce oligomerů a agregátů A? (k prevenci nástupu onemocnění), oligomerů tau a neurofibrilárních klubek (k eliminaci progrese onemocnění) a mitochondriální dysfunkce (ke snížení progrese neurodegenerace). Účinnost farmakoterapie lze zvýšit vhodnou kombinací s jinými lékovými i nelékovými intervencemi a doplňky stravy.
Klíčová slova: Alzheimerova nemoc, biomarker, léčivo, neurodegenerace, rizikový faktor
SUMMARY
Fišar Z, Hroudová J. Alzheimer´s disease: risk factors, biomarkers, and pharmacotherapeutic strategies
Knowledge about risk factors and biomarkers of Alzheimer´s disease (AD), which are the basis for understanding the pathophysiology of the disease and for identifying the cellular targets for new drugs, is summarized. The main targets of new AD drugs are processes associated with amyloid beta (A?) and tau neurotoxicity, impaired neurotransmission metabolic dysregulation, mitochondrial dysfunction, oxidative stress, neuroinflammation, neuroplasticity, proteostasis, and proteinopathy. Some new drugs target multiple associated cellular processes, such as cholinergic depletion, glutamate toxicity, A? aggregation, tau hyperphosphorylation, oxidative stress, and mitochondrial protein activity. Pharmacological intervention in AD with the potential to be causal includes targeting the production, elimination, distribution, and interactions of A? oligomers and aggregates (to prevent disease onset), tau oligomers and neurofibrillary tangles (to prevent disease progression), and mitochondrial dysfunction (to reduce neurodegeneration progression). The effectiveness of pharmacotherapy may be enhanced by appropriate combination with other drug and non-drug interventions and nutritional supplements.
Key words: Alzheimer´s disease, biomarker, drug, neurodegeneration, risk factor
ÚVOD
Ve vztahu k rizikovým faktorům, biomarkerům a klinickým příznakům lze rozlišit různé formy Alzheimerovy nemoci (AD). Hlavním rizikovým faktorem je věk. Ve většině případů je AD rozpoznána po 65. roce věku a označuje se jako sporadická forma AD s pozdním nástupem (LOAD). V 5-10 % případů dochází k časnějšímu nástupu onemocnění; tato forma AD se označuje jako AD s časným nástupem (EOAD) a je silně geneticky podmíněná.1 Genetické hledisko umožňuje rozlišit sporadickou AD (vznikající v důsledku kombinace genetiky, prostředí a životního stylu a nemající rodinnou vazbu) a autosomálně dominantní (familiární) AD (ADAD), která je definována jako dominantně dědičná AD s patologickým potvrzením a vyskytuje se v méně než 1 % všech případů. ADAD lze chápat jako podskupinu EOAD. Studium ADAD umožňuje určit časovou sekvenci změn biomarkerů u osob náchylných k rozvoji geneticky podmíněné AD.2 Předpokládá se, že všechny formy AD sdílejí podobné patofyziologické procesy, s tím, že ADAD je způsobena především amyloidopatií, zatímco u polygenních sporadických forem AD mohou k rozvoji onemocnění významně přispívat další faktory.
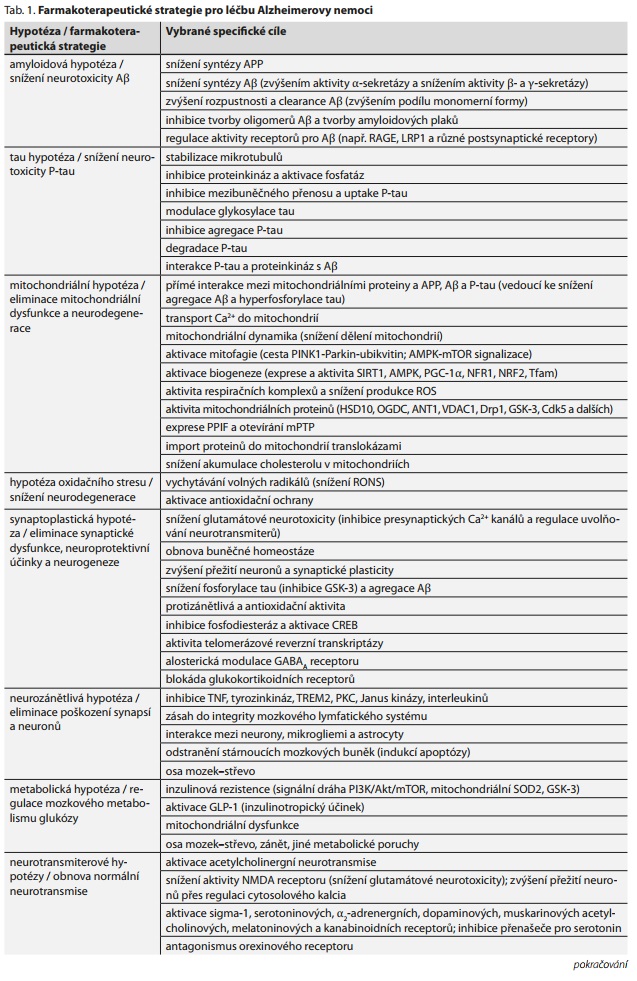
Ačkoli je etiologie AD dlouho a intenzivně studována a jsou nalézány nové biomarkery, zůstávají klíčovými hypotézami AD amyloidová a tau hypotéza a za příčiny vzniku a progrese onemocnění je považována především amyloidopatie, tauopatie, mitochondriální dysfunkce, metabolické poruchy, neurozánět a oxidační stres.3 Bylo potvrzeno, že za neuronální poškození a smrt jsou zodpovědné rozpustné oligomery A? a tau a na AD lze nahlížet také jako na oligomeropatii.4 A? oligomery spouštějí konverzi tau na toxickou oligomerní formu; zároveň může tau prostřednictvím zpětné vazby zvyšovat toxicitu AP oligomerů.5 Neurotoxicita A? a fosforylovaného tau (P-tau) je uskutečňována kaskádou procesů zahrnující neurozánět, mitochondriální dysfunkci, oxidační stres a narušení metabolických drah, které vedou k patologii a symptomům AD.3 Hlavními cíli nových potenciálních léčiv AD jsou proto procesy související s neurotoxicitou A? a P-tau, mitochondriální dysfunkcí, metabolickými poruchami a neurozánětem.
Tento přehled shrnuje poznatky o rizikových faktorech a biomarkerech AD a o farmakoterapeutických strategiích založených na poznání neurodegenerativních procesů při AD. Molekulární cíle nově vyvíjených léčiv pro AD, především chorobu modifikujících léčiv ("disease-modifying drugs", DMD), zahrnují buněčné procesy a interakce související s amyloidovou, tau a mitochondriální neurotoxicitou.
RIZIKOVÉ FAKTORY A BIOMARKERY ALZHEIMEROVY NEMOCI
Rizikové faktory jsou k onemocnění vázány kauzálně, zatímco rizikové markery mohou, ale nemusejí, být s onemocněním spojeny kauzálně. Biologické markery (biomarkery) jsou objektivně měřitelné indikátory normálních biologických procesů, patogenních procesů nebo farmakologické odpovědi na terapeutickou intervenci. Biomarkery lze považovat za podskupinu rizikových markerů, které jsou objektivně měřitelné. Lze je použít pro diagnostiku a sledování progrese a účinnosti léčby onemocnění. Poznání rizikových faktorů a biologických markerů je nezbytné pro pochopení patofyziologie AD a nalezení molekulárních cílů nových léčiv.
Rizikové faktory pomáhají identifikovat jedince, kteří jsou v budoucnu vystaveni většímu riziku rozvoje onemocnění než běžná populace. Pro AD jsou rizikovými faktory pokročilý věk, ženské pohlaví, přítomnost alely APOE?4 kódující izoformu apolipoproteinu E4 (ApoE4), další genetické a epigenetické variace, poranění mozku, environmentální faktory a stresory, včetně nízké úrovně vzdělání, životního stylu, infekce, kardiovaskulárního onemocnění a metabolických dysregulací jako diabetes mellitus 2. typu (T2DM).3,6-8 Progresi AD ovlivňuje životní styl a kognitivní rezerva, takže vývoj onemocnění může být značně individuální.
Předpokládá se, že stárnutí, genetické a epigenetické faktory spolu s vlivem rizikových faktorů vnějšího i vnitřního prostředí vedou k iniciaci rozvoje sporadické formy AD, a pro poznání primárních kauzálních příčin AD je nezbytný longitudinální výzkum rizikových faktorů a biomarkerů od počátku onemocnění do rozvoje demence (obr. 1).
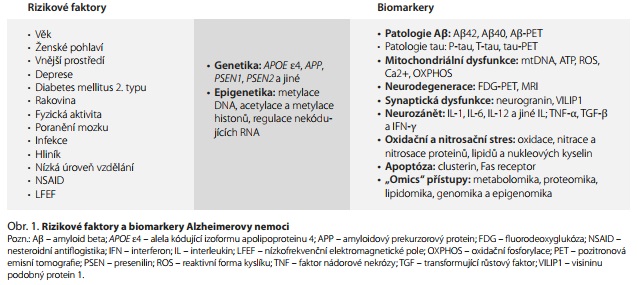
Věk
Vzhledem k tomu, že hlavním rizikovým faktorem sporadické formy AD je věk, předpokládá se, že iniciačním faktorem vzniku a rozvoje onemocnění mohou být poruchy metabolismu a bioenergetiky spojené se stárnutím. Životní riziko pro demenci a AD je vyšší u žen než u mužů a narůstá s věkem.9 Biologie stárnutí je spojena s metabolickým a oxidačním stresem, záněty, mutacemi DNA a souvisejícími procesy. Je navržena řada propojených buněčných charakteristických znaků stárnutí zahrnující nestabilitu genomu, opotřebování telomer, epigenetické změny, ztrátu proteostázy, mitochondriální dysfunkci, buněčné stárnutí, vyčerpání kmenových buněk, změněnou mezibuněčnou komunikaci, chronický zánět a dysbiózu.10 Vzhledem ke klíčové úloze mitochondrií v bioenergetice, metabolismu, neurozánětu, neuroplasticitě, oxidačním stresu a apoptóze je dlouhodobě pozornost věnována snížené mitochondriální funkci při stárnutí a neurodegeneraci. Předpokládá se, že specifická neuropatologie AD může být spuštěna nebo urychlena mitochondriální dysfunkcí spojenou se stárnutím.3,11,12
Současné strategie pro prevenci procesů spojených se stárnutím a neurodegenerací jsou zaměřeny na mechanismy kontroly kvality mitochondrií, na regulaci oxidační fosforylace, tvorby ATP, oxidačního stresu, apoptózy a autofagie a na stimulaci mitochondriální biogeneze metabolickými modulátory, léky, dietou (včetně kalorické restrikce) a cvičením.13
Rizikové faktory vnějšího prostředí
Environmentální faktory ovlivňující neurodegeneraci zahrnují kardiovaskulární onemocnění, fyzickou aktivitu, stravu, vzdělání, traumatické poranění mozku, depresi a další. Vztah těchto vlivů prostředí ke specifickým patologickým změnám u AD není dosud dostatečně prozkoumán. Nejvýznamnějšími rizikovými faktory vnějšího prostředí pro rozvoj AD jsou deprese v pozdním věku a T2DM.6 Významnými faktory jsou také rakovina, deprese v jakémkoli věku a fyzická aktivita. Méně významný je vliv hliníku, vzdělání, herpetických infekcí, nízkofrekvenčního elektromagnetického pole a nesteroidních antiflogistik. Určitý vliv může mít také alkohol, vitamin C a E, infekce Chlamydophila pneumoniae, spirochetální infekce, obezita, mírné traumatické poranění mozku a další.
Genetické rizikové faktory
Rodinná anamnéza demence značně zvyšuje riziko rozvoje AD. Studie dvojčat (monozygotních i dizygotních) potvrdila, že dědičnost AD je vysoká (58 %) a že stejné genetické faktory mají vliv na muže i ženy. Pro ADAD, resp. familiární EOAD, byly identifikovány tři kauzální geny, které kódují proteiny regulující štěpení amyloidového prekurzorového proteinu (APP) a tvorbu A?: (1) gen pro APP (APP) na chromozomu 21 (21q21.1-21q21.3), (2) gen pro presenilin 1 (PSEN1) na chromozomu 14 (14q24.3) a (3) gen pro presenilin 2 (PSEN2) na chromozomu 1 (1q31-q42).14 Nejčastější příčinou dědičnosti EOAD je mutace PSEN1; mutace PSEN2 a APP jsou méně časté. Mutace v těchto genech ale vysvětlují jen 5-10 % celkové EOAD, takže velká skupina pacientů s EOAD není dosud geneticky vysvětlena.15 Další kauzální geny se hledají pomocí technologií nové generace, jako je sekvenování celého genomu a sekvenování celého exomu.
Výskyt alely APOE ?4 (19q13.32) je hlavním genetickým rizikovým faktorem pro sporadickou LOAD.7 ApoE4 zvyšuje neurotoxicitu A? a tau při AD. Celogenomové asociační studie (GWAS) a nové genomické techniky identifikovaly mnoho možných lokusů/genů spojených s AD.16 Odhaduje se, že počet kauzálních společných jednonukleotidových polymorfismů pro LOAD může být menší než 100. Dvoustupňová GWAS se 111 326 klinicky diagnostikovanými případy AD a 677 663 kontrolami našla 75 (potvrdila 33 dřívějších a objevila 42 nových) rizikových lokusů a byla identifikována řada nových geneticky asociovaných procesů. Pro objasnění vlivu dědičnosti u AD je nezbytné zvětšení velikosti vzorku GWAS. Analýza těchto dat potvrdila zapojení amyloidových a tau drah a mikroglií do patofyziologie AD.17 Studium vzácných genetických variant spojených s AD může identifikovat nové kódující sekvence lokalizované např. v PLD3, TREM2, ABI3, PLCG2, PILRA, ABCA7 a SORL1. K identifikaci genů AD je třeba tyto genetické rizikové faktory zmapovat na varianty a geny pomocí funkčních genomických studií, což teprve probíhá.8
Fyziologická úloha genů spojených s AD zahrnuje regulaci neuronální homeostázy, metabolismu tau a AP, vápníku, lipidů, synaptické plasticity, neuronální proliferace a diferenciace, imunoregulace, stabilizace cytoskeletu a aktivity mitochondriálních komplexů. V patologických stavech jsou tyto geny spojeny s patologií tau a AP, neurozánětem, mitochondriální dysfunkcí, narušením synaptické plasticity, oxidačním stresem a narušením integrity hematoencefalické bariéry.18
Epigenetické změny (metylace a hydroxymetylace DNA, acetylace a metylace histonů a regulace nekódujících RNA) se ukázaly jako důležité v patogenezi AD. Mitochondriální DNA (mtDNA) může být také epigeneticky regulována. Vzhledem k tomu, že epigenetické změny lze detekovat i na periferii, mají potenciál být zařazeny na seznam rizikových faktorů a biomarkerů AD a stát se terapeutickými cíli nových léků.
Biomarkery
Biomarkery mají podstatnou úlohu v poznání biologických základů AD a možnostech diagnostiky a prognózy onemocnění a sledování výsledků léčby.19 Biomarkery AD zahrnují genetické, biochemické, fyziologické a neurozobrazovací parametry zapojené do neurodegenerace, narušené neuroplasticity a poškození struktury a funkce mozku.
V současnosti používané diagnostické metody jsou často nákladné a časově náročné, invazivní, špatně dostupné a nedostatečně citlivé pro detekci počátečních stadií onemocnění. Nalezení specifických, citlivých, neinvazivních a levných biomarkerů AD schopných identifikovat časný nástup onemocnění je zásadní pro účinnou a cílenou terapii. Nejslibnější jsou biomarkery odvozené ze strukturálního a funkčního zobrazení mozku a biomarkery genetické a biochemické měřené v mozko-míšním moku (CSF) nebo v periferní krvi.
Z patofyziologického hlediska jsou biomarkery AD vztaženy k amyloidové patologii (toxicita amyloidových oligomerů a plaků), tau patologii (toxicita tau oligomerů a neurofibrilárních klubek), mitochondriální dysfunkci, neurozánětu, neurodegeneraci, synaptickým změnám a funkci hematoencefalické bariéry. Pro pochopení etiologie AD je důležitá znalost časového vývoje biomarkerů. Pro klinickou praxi byl navržen systém AT(N), klasifikační schéma založené na měření biomarkerů amyloidové patologie, tau patologie a neurodegenerace.20 Tento systém lze dále rozšířit na ATX(N) klasifikaci zahrnující biomarkery mitochondriální, zánětlivé, synaptické, oxidačního stresu a vaskulární.21 K usnadnění klinické interpretace biomarkerů A? a tau v CSF lze použít Algoritmus Erlangen Score.22
Neurozobrazovací biomarkery
Neurozobrazovací biomarkery AD se zaměřují na změny strukturální (volumetrie, ztenčení kůry) a funkční (např. pomocí fMRI), na pokles konektivity a na patologické agregáty A? a tau; nověji se provádí také zobrazování změn epigenetických a změn spojených s neurozánětem a synaptickou integritou.23 Slibnými neurozobrazovacími biomarkery AD jsou objem hipokampu a entorhinální kůry, strukturální změny cholinergních jader v bazálním předním mozku a tloušťka mozkové kůry.24
Skenování pomocí pozitronové emisní tomografie (PET) s použitím specifických tracerů pro A? a tau umožňuje in vivo studium špatně složených a nahromaděných proteinů (nikoli však monomerů a oligomerů) v mozku.25 Pro monitorování ukládání A? v mozku lze použít Pittsburskou sloučeninu B značenou 11C (11C-PiB) nebo florbetapir značený 18F. Ukládání tau lze sledovat pomocí novějších látek vázajících se na tau a značených 18F. Neurodegeneraci a synaptické poškození lze kvantifikovat pomocí PET s 18F-fluorodeoxyglukózou (18F-FDG) nebo analýzou dat magnetické rezonance (MRI).24
Krevní biomarkery
Měření biomarkerů v krvi je méně invazivní a může být cenově i časově efektivnější než měření biomarkerů v CSF nebo pomocí neurozobrazovacích metod. V současnosti ale nejsou krevní biomarkery dostatečně standardizované a validované pro použití v klinické praxi. Vhodnými krevními biomarkery se jeví plazmatický P-tau217, P-tau181 a poměr A?42/A?40.26, 27 Mezi slibné krevní biomarkery AD patří především ty, které jsou vztaženy k apoptóze a neurodegeneraci, jako je klusterin, a panely biomarkerů získané pomocí pokročilých spektroskopických a "omických" metod, imunoanalýzy a hmotnostní spektrometrie.28
Proteomické a metabolomické studie poskytly panely potenciálních biomarkerů zahrnující kyselinu arachidonovou, N,N-dimetylglycin, thymin, glutamin, kyselinu glutamovou a cytidin, které umožňují odlišení pacientů s AD od nedementních kontrol a od jiných typů demence.29 Při interpretaci biomarkerů AD měřených v periferní krvi je nutné brát v úvahu účinky podávaných léků a komorbidní onemocnění, jako je deprese nebo infekce.
Jako mitochondriální biomarkery jsou studovány v mozku i krvi změny v mtDNA, produkci ATP a systému oxidační fosforylace (OXPHOS), produkci reaktivních forem kyslíku (ROS), nitrobuněčné kalciové homeostázi a uvolňování apoptotických faktorů. Do panelu krevních biomarkerů AD lze zařadit sníženou aktivitu mitochondriálního komplexu IV, mitochondriální bazální respiraci a maximální kapacitu elektronového transportního systému měřené v intaktních krevních destičkách.30
Na patogenezi AD se podílejí různé cytokiny, včetně interleukinů (IL-1, IL-6, IL-12 a další rodiny), faktor nádorové nekrózy alfa (TNF-?), transformující růstový faktor beta (TGF-?) a interferon gama (IFN-?). Potenciální nové biomarkery AD zahrnují neurogranin a protein podobný visininu 1 (VILIP-1) jakožto biomarkery synaptické a neuronální degenerace AD, lehký polypep-tid neurofilamentů (NfL), jakožto tau-nezávislý marker neuroaxonální degenerace, kyselý gliový fibrilární protein (GFAP), jakožto plazmatický biomarker AD, a rozpustnou formu spouštěcího receptoru exprimovaného na myeloidních buňkách 2 (TREM2), jakožto biomarker aktivace mikroglií schopný vázat ApoE.3
Slibné a validované biomarkery
Jako slibné biomarkery AD jsou studovány (i) v CSF: A?42, celkový tau (T-tau), P-tau a tau/A?42; (ii) v periferní krvi: T-tau, A?42/A?40 a NfL; (iii) v mozku: objem levého a pravého hipokampu, objem entorhinální kůry, atrofie mediálního temporálního laloku, 18F-FDG PET, 11C-PiB PET; a (iv) APOE ?4.27 Za dostatečně validované pro klinickou diagnostiku a sledování účinků terapie se považují pouze dva biomarkery amyloidózy (snížený A?42 v CSF a zobrazení amyloidu v mozku pomocí PET) a biomarkery neurodegenerace (zvýšený T-tau a P-tau, NfL a VILIP-1 v CSF, atrofie měřená pomocí strukturální MRI a hypometabolismus měřený pomocí PET s 18F-FDG).31 V některých klinických zkouškách nových potenciálních léčiv pro AD se pro sledování jejich účinků používá také měření GFAP v CSF a plazmě a elektroencefalografie. Biomarkery oxidačního stresu v mozku osob s AD byly dobře zdokumentovány pomocí markerů pro oxidaci proteinů, lipidů a nukleových kyselin.32
Časový průběh abnormalit biomarkerů
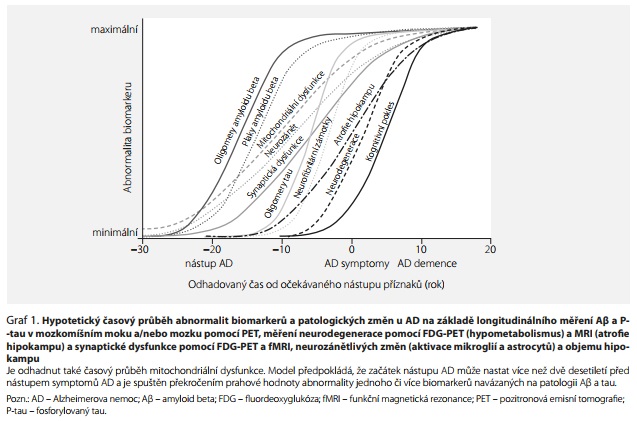
Z klinického hlediska lze vývoj AD rozdělit do tří stadií: (1) preklinická AD charakterizovaná jako asymptomatická cerebrální amyloidóza; (2) AD s průkazem A? spolu s důkazem synaptické dysfunkce nebo časné neurodegenerace; a (3) AD s průkazem A? spolu s důkazem neurodegenerace a mírné kognitivní poruchy (MCI) a demence v důsledku AD. Stadia demence v důsledku AD se dělí na mírnou, střední a těžkou demenci.33,34 Zatímco identifikace a posouzení progrese patologie AD je post mortem založena na stagingových systémech,35,36 u živých osob je založena na měření biomarkerů.19
Z výzkumného hlediska se pozornost věnuje procesům spojeným se stárnutím a jejich ovlivnění genetickými faktory a vnějším i vnitřním prostředím, resp. studiu patologických procesů v mozku mnoho let před klinicky rozpoznaným nástupem onemocnění. Byla navržena sekvence biomarkerů pro predikci rizika progrese onemocnění k MCI a demenci založená především na amyloidové a tau hypotéze AD.19,34
Kohortová studie ukládání A?, neurodegenerace a kognitivního poklesu u sporadické AD potvrdila, že akumulace A? v mozku související se snížením objemu hipokampu a šedé hmoty mozkové a zhoršením paměti pravděpodobně trvá více než dvě desetiletí.37 Další longitudinální studie kognitivních funkcí, biomarkerů v CSF a zobrazovacích biomarkerů založených na MRI a PET s 11C-PiB a 18F-FDG potvrdily, že PiB-PET je asociována s AD, neuritickými plaky a difuzními plaky a FDG-PET a MRI korelují s neuropatologickými nálezy u AD.38 Přinejmenším u části pacientů s AD předchází akumulace A? následnou akumulaci tau.5,25 Předpokládá se, že toxicita rozpustných A? oligomerů může být primárně zodpovědná za nástup onemocnění, zatímco rozpustné tau oligomery a neurofibrilární klubka (NFT) mohou být zodpovědné za progresi neurodegenerace při AD.39 Model, který považuje časnou akumulaci A? a P-tau za primární události v patofyziologické kaskádě AD, nelze považovat za zcela potvrzený a obecně platný, protože existuje řada procesů, které mohou předcházet spuštění patologie A? a P-tau, jako jsou mitochondriální, metabolické, neurozánětlivé, cytoskeletální a další neuronové alterace související s věkem.
Model dynamických biomarkerů AD, tj. závislost velikosti abnormalit biomarkerů na čase a klinickém stadiu AD, je podložen longitudinálním měřením (i) amyloidové patologie (A?42 v CSF a zobrazení AP v mozku pomocí PET), (ii) tau patologie (P-tau a T-tau v CSF), (iii) neurodegenerace (struktura mozku volumetrickou MRI) a synaptické dysfunkce (FDG-PET a fMRI),2,25,34,40 (iv) neurozánětlivých změn, jako je aktivace mikroglií a astrocytů,41 a (v) klinických parametrů, jako je kognitivní deficit a stadium demence (graf 1). Nositelé alely APOE ?4 přitom mohou mít dřívější nástup kaskády patofyziologických procesů iniciované A?.19,34 Vzhledem ke klíčové úloze mitochondrií v regulaci buněčných funkcí se jako vhodné jeví zařazení mitochondriální dysfunkce související s věkem do tohoto modelu dynamických biomarkerů (založeného především na amyloidové a tau hypotéze). Poznání časování negenetických biomarkerů pomáhá určit příčiny vzniku a progrese AD. Neurotoxicita A? a tau oligomerů, mitochondriální dysfunkce, neurozánět a metabolické poruchy se jeví jako nejvhodnější kandidáti na primární spouštěč nástupu AD.
Hypotetický časový průběh měřitelných patofyziologických parametrů ve vztahu ke klinickému průběhu AD lze použít jako základ pro vývoj nových léků proti AD zacílených na patofyziologické procesy v časných stadiích onemocnění. Vzhledem k provázanosti mozkových procesů vedoucích k neurodegeneraci při AD (neurotoxicita A? oligomerů a neuritických plaků, neurotoxicita tau oligomerů a NFT, mitochondriální dysfunkce a neurozánětlivé procesy spojené s aktivací mikroglií a astrocytů) a pozorování, že doba mezi vznikem onemocnění a rozpoznáním kognitivního deficitu (preklinická AD) může činit až 20 nebo více let, má zřejmě smysl cílit farmakoterapii na různé systémy regulující neurodegeneraci, neuroplasticitu a neurochemii mozku.
FARMAKOTERAPEUTICKÉ STRATEGIE PRO ALZHEIMEROVU NEMOC
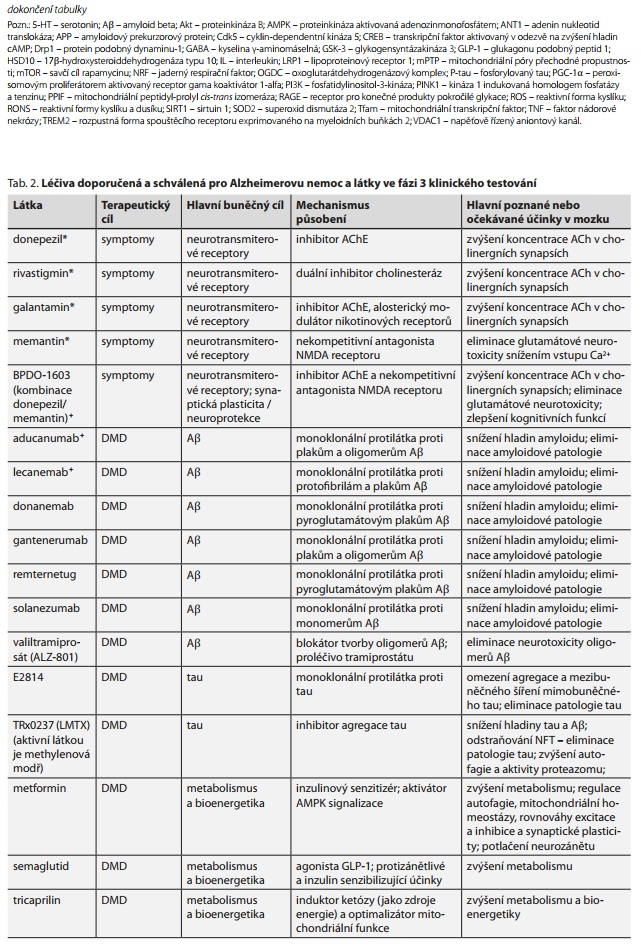
Léky schválené nebo doporučené v současné době pro léčbu AD patří do kategorie látek cílících na neurotransmiterové receptory (inhibitory cholinesteráz a antagonista N-metyl-d-aspartátového (NMDA) receptoru) a A? (monoklonální protilátky namířené proti plakům, protofibrilám a oligomerům A0). Delší dobu jsou v klinickém využití léky cílené na potlačení či zmírnění symptomů AD (donepezil, rivastigmin, galantamin, memantin a kombinace memantin/donepezil). Nově získala urychlené schválení Úřadu pro kontrolu potravin a léčiv Spojených států amerických (FDA) dvě chorobu modifikující léčiva (DMD) cílená na patologii AP (aducanumab a lecanemab).
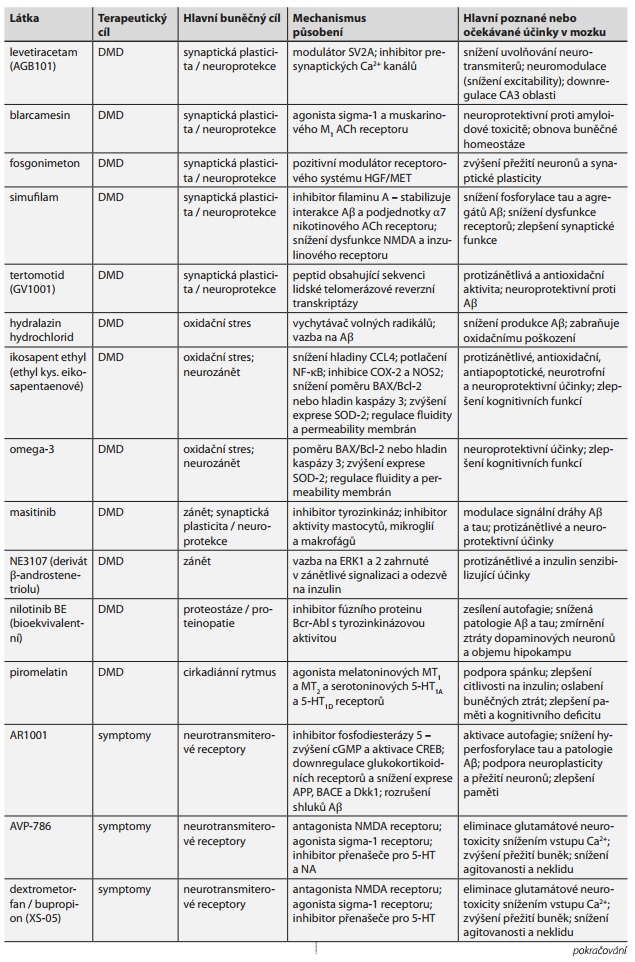
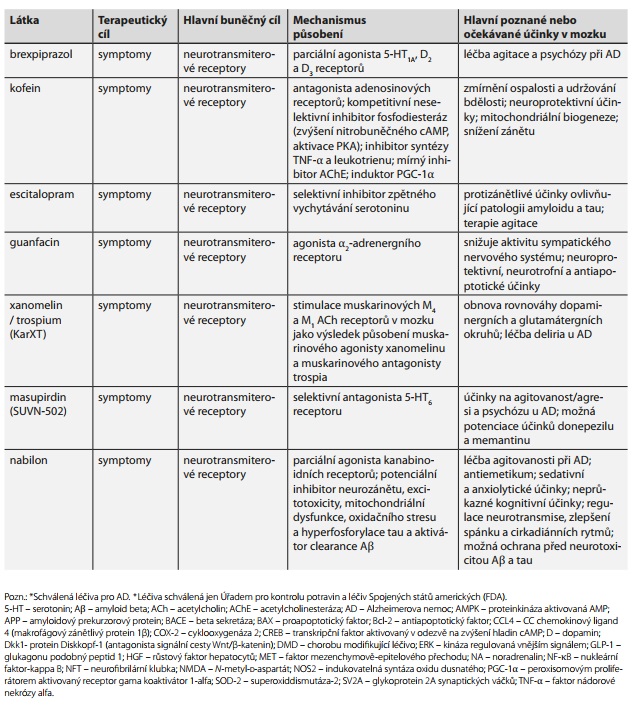
Cíle léčiv
Molekulární cíle nově vyvíjených léčiv jsou dány poznáním patofyziologie, rizikových faktorů a biomarkerů AD. Strategie vývoje nových léčiv pro AD akceptuje skutečnost, že pro účinnou terapii AD je nutné zahájit terapeutickou intervenci již v časném stadiu onemocnění a využít multifaktoriální přístup zohledňující jednotlivé iniciátory rozvoje AD. Pozornost se věnuje především vývoji nových účinných a specifických DMD. Probíhá ale také testování symptomatických látek cílených na zlepšení kognitivních funkcí a na neuropsychiatrické symptomy při AD, přičemž se často jedná o léky již schválené pro léčbu jiných onemocnění nebo látky používané v alternativní a komplementární medicíně.42
Projekty, které jsou zaměřené na identifikaci, validaci a vývoj potenciálních cílů pro terapeutickou intervenci při AD a hodnocení terapeutických účinků látek v různých fázích klinického testování (včetně nefarmakologické intervence), používají klasifikační systém CADRO ("Common Alzheimer´s and Related Dementias Research Ontology"; Ontologie výzkumu Alzheimerovy choroby a souvisejících demencí; https://iadrp.nia.nih.gov/about/cadro) vyvinutý Národním institutem pro stárnutí ("National Institute on Aging"; NIA; https://www.nia.nih.gov/) a Alzheimerovskou asociací ("Alzheimer´s Association"; https://www.alz.org/). CADRO rozlišuje tyto kategorie biologických procesů a cílů léčiv: A?; tau; ApoE, lipidy a lipoproteinové receptory; neurotransmiterové receptory; neurogeneze; zánět; oxidační stres; buněčná smrt; proteostáze/proteinopatie; metabolismus a bioenergetika; vaskulatura; růstové faktory a hormony; synaptická plasticita/neuroprotekce; osa střevo-mozek; cirkadiánní rytmus; environmentální faktory; epigenetické regulátory; vícecílová léčiva; a jiné.
Přesnější specifikaci cílů léčiv pro AD umožňuje znalost buněčných procesů, které jsou propojeny a vedou k nástupu onemocnění a jeho progresi. Neurodegenerace, synaptická dysfunkce a ztráta neuronů je složitou kaskádou buněčných procesů, která zahrnuje patologii oligomerů a plaků A?, patologii oligomerů tau a NFT, neurozánět, mitochondriální dysfunkci, metabolickou dysregulaci a deficit degradace proteinů. Specifické účinky oligomerů A? a P-tau mohou být navíc regulovány tau, APP a inzulinovou rezistencí mozku.3 Farmakoterapeutické strategie pro léčbu AD plynoucí z těchto poznatků jsou shrnuty v tab. 1. Lze očekávat, že snížení neurotoxicity A? lze dosáhnout především přes (i) snížení syntézy APP, (ii) snížení syntézy A? (zvýšením aktivity ?-sekretázy a snížením aktivity ?? a ??sekretázy), (iii) zvýšení clearance A? (např. zvýšením podílu monomerní formy), (iv) inhibici tvorby oligomerů A? a tvorby amyloidových plaků, a (v) regulaci receptorů pro A?, např. receptoru pro konečné produkty pokročilé glykace (RAGE), lipoproteinového receptoru 1 (LRP1) a různých postsynaptických receptorů.3
Testovaná léčiva
Podle pravidelného ročního přehledu vývoje léčiv pro AD43 založeného na analýze dat z ClinicalTrials.gov bylo na začátku roku 2023 v klinických studiích ve fázi 1, 2 nebo 3 celkem 141 látek testovaných pro léčbu AD a mírné kognitivní poruchy (MCI) související s AD. Ve fázi 3 klinických studií se nachází 36 látek, ve fázi 2 je 87 látek a ve fázi 1 je 31 látek. Testované látky jsou většinou zaměřené (i) na změnu patofyziologie AD, tj. látky typu chorobu modifikujících léčiv (DMD, 78.7 %), (ii) na neuropsychiatrické symptomy (10,6 %) a (iii) kognitiva a nootropika ("cognitive enhancers", 10,6 %). Nejčastějšími cíli jsou zánět (17,0 %), A? (15,6 %), synaptická plasticita/neuroprotekce (12,9 %), tau (9,2 %), metabolismus a bioenergetika (7,1 %) a oxidační stres (5,0 %). Na neurotransmiterové receptory cílí více než 20 % testovaných látek, přičemž se jedná vesměs o léčiva cílící na potlačení neuropsychiatrických symptomů a zvýšení kognitivních funkcí při AD. Zvyšující se příklon ke studiu DMD potvrzuje změna v počtu látek testovaných ve fázi 3, 2 a 1: zatímco DMD tvoří 66,7 % (24 z 36) látek ve fázi 3, 85,1 % (74 z 87) látek ve fázi 2 a 80,6 % (25 z 31) látek ve fázi 1, tak pro látky cílené na neurotransmiterové receptory to je 30,6 % (11 z 36), 13,8 % (12 z 87) a 19,4 % (6 z 31) látek. Z látek testovaných pro léčbu AD patří 28 % mezi léky již schválené pro jiné terapie. Schválená nebo doporučená léčiva pro AD a látky testované ve fázi 3 jsou uvedeny v tab. 2 spolu s jejich molekulárními cíli a mechanismem působení.43
Mnoho perspektivních látek pro léčbu nebo přídavnou léčbu AD je v testovací fázi 2.43 Z látek, jejichž terapeutickým cílem je DMD, je to např. L-serin, inzulin a řada monoklonálních protilátek proti A? a tau. Na zlepšení kognitivních funkcí nebo zpomalení progrese AD může mít vliv např. nikotin, trazodon, nebo tradiční čínská medicína. Na potlačení neuropsychiatrických symptomů při AD se mohou podílet např. kanabinoidy, dronabinol a kanabidiol. Neurochemie a neuroplasticita mozku může být ovlivněna také životním stylem, vhodnou léčbou komorbidních onemocnění, psychosociálními intervencemi a dietou.
Kromě nových DMD léčiv cílících na primární příčiny nástupu a progrese AD se hledají a testují také vhodné kombinace schválených léčiv s adjuvantními látkami. Tyto volně prodejné doplňky (jako w-3 mastné kyseliny, sója, ginkgo biloba, vitamíny B, vitamín D plus vápník, vitamín C nebo ?-karoten) nemají samy o sobě dosud prokázané účinky na prevenci kognitivních dysfunkcí při AD.44
Molekulární a buněčné mechanismy účinku adjuvantních látek jsou komplexní a nejsou dobře známé. Lze očekávat, že pokud budou mít účinky antiamyloidní, anti-tau, neurochemické, mitochondriální, antioxidační nebo protizánětlivé, mohou mít také terapeutický potenciál ke zmírnění progrese kognitivní poruchy při AD. Na inhibici neurotoxicity A? se mohou podílet např. resveratrol, huperzin A a karvakrol (z dobromysli). Agregaci a toxicitu P-tau mohou zřejmě modulovat červený ženšen, krocin (šafránová žluť), cinnamaldehyd (ze skořicové kůry) a epikatechin (ze zeleného čaje), purpurin a folát.3 Vliv organofosforových sloučenin, přírodních produktů, indické a čínské medicíny a dalších adjuvantních látek na kognitivní deficit u AD ale stále není prokázán.
Antiamyloidová imunoterapie je založena na snížení hladin amyloidu v mozku a s tím spojenou modifikací průběhu onemocnění. Aducanumab a lecanemab byly urychleně schváleny FDA, další monoklonální protilátky snižující patologii A? jsou testovány ve fázi 3 klinického testování (tab. 2). Účinnost antiamyloidové imunoterapie podporuje amyloidovou hypotézu AD; její vliv na progresi kognitivního poklesu nebo demence u AD ještě musí být potvrzen.45 Nedostatečná účinnost protilátek a vakcín proti A? při terapii kognitivního poklesu ukazuje, že je nutné zabránit již časné tvorbě A? a tau oligomerů. Možnost perorálního podávání a mechanismus působení založený na inhibici tvorby oligomerů AP má např. valiltramiprosát (ALZ-801; proléčivo homotaurinu).46 Také anti-tau terapie nebyly dosud dostatečně úspěšné a jsou předmětem dalšího výzkumu.47
Dosud schválená DMD léčiva pro AD podporují amyloidovou hypotézu, ale buněčné cíle nových klinicky testovaných DMD jsou vztaženy také k hypotéze oxidačního stresu, tau, mitochondriální, metabolické, neurozánětlivé, nebo synaptoplastické hypotéze. Testovaná léčiva potlačující symptomy onemocnění jsou většinou vztažena k neurotransmiterové hypotéze. Rozvoj, propojování či sjednocování hypotéz AD a poznání časového průběhu abnormalit biomarkerů pomáhá upřesnit perspektivní cíle nových DMD. Pravděpodobná úloha patologie A?, tau a mitochondriální dysfunkce v iniciaci AD, spolu se vzájemnou interakcí a potenciací těchto procesů,3 ukazuje na perspektivu zacílení nových léčiv na neurotoxicitu A? a tau oligomerů a s ní spojenou narušenou bioenergetiku, metabolismus a neurozánět. Slibným terapeutickým cílem nových kauzálních léků proti AD je eliminace jak tvorby a šíření, tak mitochondriální toxicity A? oligomerů a P-tau oligomerů. Při vývoji nových léčiv pro nemoci s komplexním mechanismem patogeneze, jako je AD, se často testují ligandy zaměřené na více cílů ("multitarget-directed ligand", MTDL), které jsou schopny zasáhnout a modulovat několik různých interagujících cílů na různých úrovních.48
ZÁVĚR
Molekulární cíle nových léčiv pro AD vycházejí z poznání (i) mechanismů účinků již schválených léčiv, (ii) biomarkerů a příčin vzniku a progrese onemocnění a (iii) mechanismů stárnutí a neurodegenerace. Klíčovou úlohou je poznání rizikových faktorů a biomarkerů souvisejících s neurotoxicitou A? a tau, metabolickými poruchami, mitochondriální dysfunkcí, oxidačním stresem, neurozánětem a narušením synaptické i strukturální plasticity. Nově testované MTDL cílí především na snížení cholinergní deplece, toxicitu glutamátu, agregaci A?, hyperfosforylaci tau, oxidační stres a aktivitu mitochondriálních proteinů. Účinnost farmakoterapie lze zvýšit vhodnou kombinací s jinými lékovými i nelékovými intervencemi a doplňky stravy, které mohou mít příznivé účinky na mozkové funkce. Jako perspektivní se jeví zaměření se na interakce a zpětnovazebné regulace mezi oligomery A?, oligomery P-tau a mitochondriemi. Tyto signální dráhy a interakce však musejí být dále podrobně prozkoumány z hlediska jejich úlohy v neurodegeneraci a progresi AD.
Použité zkratky: 5-HT - serotonin; A? - amyloid beta; ACh - acetylcholin; AChE - acetylcholinesteráza; AD - Alzheimerova nemoc; ADAD - autosomálně dominantní (familiární) AD; Akt - proteinkináza B; AMPK - proteinkináza aktivovaná AMP; ANT1 - adenin nukleotid translokáza; ApoE - apolipoprotein E; APOE ?4 - alela kódující izoformu apolipoproteinu 4; APP - amyloidový prekurzorový protein; BACE - beta sekretáza; BAX - proapoptotický faktor; Bcl-2 - antiapoptotický faktor; CADRO - ontologie výzkumu AD a souvisejících demencí; CCL4 - CC chemokinový ligand 4 (makrofágový zánětlivý protein 1?); Cdk5 - cyklin-dependentní kináza 5; COX-2 - cyklooxygenáza 2; CREB - transkripční faktor aktivovaný v odezvě na zvýšení hladin cAMP; CSF - mozkomíšní mok; Dkk1- protein Diskko-pf-1; DMD - chorobu modifikující léčivo; Drp1 - protein podobný dynaminu-1; EOAD - AD s časným nástupem; ERK - kináza regulovaná vnějším signálem; FDA - úřad pro kontrolu potravin a léčiv Spojených států amerických; FDG - fluorodeoxyglukóza; fMRI - funkční magnetická rezonance; GABA - kyselina ??aminomáselná; GLP-1 - glukagonu podobný peptid 1; GFAP - kyselý gliový fibrilární protein; GSK-3 - glykogensyntázakináza 3; GWAS - celogenomová asociační studie; HGF - růstový faktor hepatocy-tů; HSD10 - 17?-hydroxysteroiddehydrogenáza typu 10; IFN - interferon; LFEF - nízkofrekvenční elektromagnetické pole; LOAD - sporadická forma AD s pozdním nástupem; LRP1 - lipoproteinový receptor 1; mtDNA - mito-chondriální DNA; MCI - mírná kognitivní porucha; MET - faktor mezenchy-mově-epitelového přechodu; mPTP - mitochondriální pór přechodné propustnosti; MTDL - ligand zaměřený na více cílů; mTOR - savčí cíl rapamycinu; NA - noradrenalin; NF-?B - nukleární faktor-kappa B; NfL - lehký polypeptid neurofilamentů; NFT - neurofibrilární klubka; NMDA - N-metyl-D-aspartát; NOS2 - indukovatelná syntáza oxidu dusnatého; NRF - jaderný respirační faktor; NSAID - nesteroidní antiflogistika; OGDC - oxoglutarátdehydroge-názový komplex; OXPHOS - oxidační fosforylace; PET - pozitronová emisní tomografie; PGC-1a - peroxisomovým proliferátorem aktivovaný receptor gama koaktivátor 1-alfa; PI3K - fosfatidylinositol-3-kináza; PiB - Pittsburská sloučenina B; PINK1 - kináza 1 indukovaná homologem fosfatázy a tenzinu; PKC - proteinkináza C; PPIF - mitochondriální peptidyl-prolyl cis-trans izomeráza; PSEN - presenilin; P-tau - fosforylovaný tau; RAGE - receptor pro konečné produkty pokročilé glykace; RONS - reaktivní formy kyslíku a dusíku; ROS - reaktivní formy kyslíku; SIRT1 - sirtuin 1; SOD2 - superoxid dismutáza 2; SV2A - glykoprotein 2A synaptických váčků; T2DM - diabetes mellitus 2. typu; TNF-? - faktor nádorové nekrózy alfa; Tfam - mitochondriální transkripční faktor; TGF - transformující růstový faktor; TREM2 - rozpustná forma spouštěcího receptoru exprimovaného na myeloidních buňkách 2; T-tau - celkový tau; VDAC1 - napěťově řízený aniontový kanál; VILIP1 - visininu podobný protein 1
LITERATURA
- 1. Almkvist O, Nordberg A. A biomarker-validated time scale in years of disease progression has identified early-and late-onset subgroups in sporadic Alzheimer´s disease. Alzheimers Res Ther 2023; 15 (1): 89.
- 2. Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC et al. Clinical and biomarker changes in dominantly inherited Alzheimer´s disease. N Engl J Med 2012; 367 (9): 795-804.
- 3. Fišar Z. Linking the amyloid, tau, and mitochondrial hypotheses of Alzheimer´s disease and identifying promising drug targets. Biomolecules 2022; 12 (1676): 1-43.
- 4. Cline EN, Bicca MA, Viola KL, Klein WL. The amyloid-beta oligomer hypothesis: beginning of the third decade. J Alzheimers Dis 2018; 64 (s1): S567-S610.
- 5. Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014; 71 (4): 505-508.
- 6. Bellou V, Belbasis L, Tzoulaki I, Middleton LT, Ioannidis JPA, Evangelou E. Systematic evaluation of the associations between environmental risk factors and dementia: An umbrella review of systematic reviews and meta-analy-ses. Alzheimers Dement 2017; 13 (4): 406-418.
- 7. Fenclová E, Albrecht J, Harsa P, Jirák R. Risk factors for Alzheimer´s disease. Čes a slov Psychiat 2020, 116 (2): 5965.
- 8. Andrews SJ, Fulton-Howard B, Goate A. Interpretation of risk loci from genome-wide association studies of Alzheimer´s disease. Lancet Neurol 2020; 19 (4): 326-335.
- 9. Perera G, Pedersen L, Ansel D, Alexander M, Arrighi HM, Avillach P et al. Dementia prevalence and incidence in a federation of European Electronic Health Record databases: The European Medical Informatics Framework resource. Alzheimers Dement 2018; 14 (2): 130-139.
- 10. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell 2023; 186 (2): 243-278.
- 11. Weidling IW, Swerdlow RH. Mitochondria in Alzheimer´s disease and their potential role in Alzheimer´s proteostasis. Exp Neurol 2020; 330: 113321.
- 12. Fišar Z, Hroudová J, Zvěřová M, Jirák R, Raboch J, Kitzlerová E. Age-dependent alterations in platelet mitochondrial respiration. Biomedicines 2023; 11 (6).
- 13. Lee D, Jo MG, Kim SY, Chung CG, Lee SB. Dietary antioxidants and the mitochondrial quality control: their potential roles in Parkinson´s disease treatment. Antioxidants (Basel) 2020; 9 (11).
- 14. Dai MH, Zheng H, Zeng LD, Zhang Y. The genes associated with early-onset Alzheimer´s disease. Oncotarget 2018; 9 (19): 15132-15143.
- 15. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer´s disease revisited. Alzheimers Dement 2016; 12 (6): 733-748.
- 16. Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer´s disease risk. Nat Genet 2019; 51 (3): 404-413.
- 17. Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N et al. New insights into the genetic etiology of Alzheimer´s disease and related dementias. Nat Genet 2022; 54 (4): 412-436.
- 18. Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P, Vargas-Rodriguez I, Cadena-Suarez AR, Sanchez-Garibay C et al. Alzheimer´s Disease: An updated overview of its genetics. Int J Mol Sci 2023; 24 (4).
- 19. Jack CR, Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer´s disease. Alzheimers De-ment2018; 14(4): 535-562.
- 20. Jack CR, Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016; 87 (5): 539-547.
- 21. Hampel H, Cummings J, Blennow K, Gao P, Jack CR, Jr., Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol 2021; 17 (9): 580-589.
- 22. Baldeiras I, Santana I, Leitao MJ, Vieira D, Duro D, Mroczko B et al. Erlangen Score as a tool to predict progression from mild cognitive impairment to dementia in Alzheimer´s disease. Alzheimers Res Ther 2019; 11 (1): 2.
- 23. Marquez F, Yassa MA. Neuroimaging Biomarkers for Alzheimer´s Disease. Mol Neurodegener 2019; 14 (1): 21.
- 24. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr., Kawas CH et al. The diagnosis of dementia due to Alzheimer´s disease: recommendations from the National Institute on Aging-Alzheimer´s Association workgroups on diagnostic guidelines for Alzheimer´s disease. Alzheimers Dement 2011; 7 (3): 263-269.
- 25. Leuzy A, Chiotis K, Lemoine L, Gillberg PG, Almkvist O, Rodriguez-Vieitez E et al. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol Psychiatry 2019; 24 (8): 11121134.
- 26. Leuzy A, Mattsson-Carlgren N, Palmqvist S, Janelidze S, Dage JL, Hansson O. Blood-based biomarkers for Alzheimer´s disease. EMBO Mol Med 2022; 14 (1): e14408.
- 27. Li RX, Ma YH, Tan L, Yu JT. Prospective biomarkers of Alzheimer´s disease: A systematic review and meta-analysis. Ageing Res Rev 2022; 81: 101699.
- 28. Park SA, Jang YJ, Kim MK, Lee SM, Moon SY. Promising blood biomarkers for clinical use in Alzheimer´s disease: a focused update. J Clin Neurol 2022; 18 (4): 401-409.
- 29. Wang G, Zhou Y, Huang FJ, Tang HD, Xu XH, Liu JJ et al. Plasma metabolite profiles of Alzheimer´s disease and mild cognitive impairment. J Proteome Res 2014; 13 (5): 2649-2658.
- 30. Fišar Z, Hansíková H, Křížová J, Jirák R, Kitzlerová E, Zvěřová M et al. Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer´s disease and depressive disorder. Mitochondrion 2019; 48: 6777.
- 31. Leuzy A, Ashton NJ, Mattsson-Carlgren N, Dodich A, Boccardi M, Corre J et al. 2020 update on the clinical validity of cerebrospinal fluid amyloid, tau, and phospho-tau as biomarkers for Alzheimer´s disease in the context of a structured 5-phase development framework. Eur J Nucl Med Mol Imaging 2021; 48 (7): 2121-2139.
- 32. Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer´s disease and mild cognitive impairment. Free Radic Biol Med 2007; 43 (5): 658-677.
- 33. Gaugler J, James B, Johnson T, Reimer J, Solis M, Weuve J et al. 2022 Alzheimer´s disease facts and figures. Alzheimers & Dementia 2022; 18 (4): 700-789.
- 34. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM et al. Toward defining the preclinical stages of Alzheimer´s disease: recommendations from the National Institute on Aging-Alzheimer´s Association workgroups on diagnostic guidelines for Alzheimer´s disease. Alzheimers Dement 2011; 7 (3): 280-292.
- 35. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82 (4): 239-259.
- 36. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58 (12): 1791-1800.
- 37. Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer´s disease: a prospective cohort study. Lancet Neurol 2013; 12 (4): 357-367.
- 38. Lowe VJ, Lundt ES, Albertson SM, Przy-belski SA, Senjem ML, Parisi JE et al. Neuroimaging correlates with neuropathologic schemes in neurodegenerative disease. Alzheimers Dement 2019; 15 (7): 927-939.
- 39. Barthelemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer´s disease. Nat Med 2020; 26 (3): 398-407.
- 40. Meftah S, Gan J. Alzheimer´s disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression. Front Synaptic Neurosci 2023; 15: 1129036.
- 41. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell 2019; 179 (2): 312-339.
- 42. Kleinová L, Cerman J, Hlávka J, Hort J. New pharmacological options in the treatment of Alzheimer´s disease (Nové farmakologické možnosti v léčbě Alzheimerovy nemoci). Cesk Slov Neurol N 2022; 85 (6): 462-469.
- 43. Cummings J, Zhou Y, Lee G, Zhong K, Fonseca J, Cheng F. Alzheimer´s disease drug development pipeline 2023. Alzheimers Dement (N Y) 2023; 9 (2): e12385.
- 44. Butler M, Nelson VA, Davila H, Ratner E, Fink HA, Hemmy LS et al. Over-the-counter supplement interventions to prevent cognitive decline, mild cognitive impairment, and clinical Alzheimer-type dementia: a systematic review. Ann Intern Med 2018; 168 (1): 52-62.
- 45. Haddad HW, Malone GW, Comar-delle NJ, Degueure AE, Poliwoda S, Kaye RJ et al. Aduhelm, a novel anti-amyloid monoclonal antibody, for the treatment of Alzheimer´s disease: a comprehensive review. Health Psychol Res 2022; 10 (3): 37023.
- 46. Tolar M, Hey J, Power A, Abushakra S. Neurotoxic soluble amyloid oligomers drive Alzheimer´s pathogenesis and represent a clinically validated target for slowing disease progression. Int J Mol Sci 2021; 22 (12).
- 47. Wang L, Bharti, Kumar R, Pavlov PF, Winblad B. Small molecule therapeutics for tauopathy in Alzheimer´s disease: walking on the path of most resistance. Eur J Med Chem 2021; 209: 112915.
- 48. Kumar B, Thakur A, Dwivedi AR, Kumar R, Kumar V. Multi-Target-Directed Ligands as an effective strategy for the treatment of Alzheimer´s disease. Curr Med Chem 2022; 29 (10): 17571803.