Česká a slovenská psychiatrie

Časopis
Psychiatrické společnosti ČLS JEP
a Psychiatrickej spoločnosti SLS
souborný článek / review article
- AKTUÁLNÍ ČÍSLO
- ARCHIV
- VYHLEDÁVÁNÍ
- PERIODIKUM
- REDAKČNÍ RADA
- PŘEDPLATNÉ
- INZERCE
- AKTUALIZOVANÉ
POKYNY
PRO AUTORY
- ISSN 1212-0383




- © Česká a Slovenská psychiatrie 2025
- © Galén 2025
ALZHEIMEROVA NEMOC: PATOFYZIOLOGIE A HYPOTÉZY
ALZHEIMER´S DISEASE: PATHOPHYSIOLOGY AND HYPOTHESES
Zdeněk Fišar, Jana Hroudová
Psychiatrická klinika 1. LF UK a VFN v Praze
Práce byla podpořena grantem AZV MZ ČR NU23-04-00032.
SOUHRN
Fišar Z, Hroudová J. Alzheimerova nemoc: patofyziologie a hypotézy
Jsou shrnuty biologické hypotézy Alzheimerovy nemoci (AD) založené na poznatcích o patofyziologii AD, které slouží jako východiska pro farmakoterapeutické strategie. Pokroky v poznání úlohy rizikových faktorů a biomarkerů AD v neurodegenerativních procesech umožňují jednak upřesnění stávajících hypotéz nebo tvorbu nových, jednak zlepšení diagnostiky onemocnění, sledování progrese onemocnění a nalezení molekulárních cílů nových léčiv. Kromě amyloidové a tau hypotézy, resp. patologie amyloidu beta a tau proteinu, se při vývoji nových léčiv a studiu mechanismů jejich účinků vychází především z hypotézy mitochondriální, metabolické, neurozánětlivé, oxidačního stresu, synaptoplastické a neurotransmiterové. Významné pro pochopení patofyziologie AD se jeví poznatky o vzájemném propojení, interakcích a synergiích buněčných patologických procesů považovaných za klíčové v hypotézách AD.
Klíčová slova: Alzheimerova nemoc, amyloid beta, biomarker, hypotéza, metabolismus, mitochondriální dysfunkce, neuroplasticita, neurotransmise, neurozánět, tau
SUMMARY
Fišar Z, Hroudová J. Alzheimer´s disease: pathophysiology and hypotheses
Biological hypotheses of Alzheimer´s disease (AD) based on knowledge about the pathophysiology of AD are summarized, which serve as starting points for pharmacotherapeutic strategies. Advances in the knowledge of the role of risk factors and biomarkers of AD in neurodegenerative processes allow the refinement of existing hypotheses or the generation of new ones, as well as the improvement of disease diagnostics, the monitoring of disease progression, and the discovery of molecular targets for new drugs. In addition to the amyloid and tau hypothesis, respectively the pathology of amyloid beta and tau protein, the mitochondrial, metabolic, neuroinflammatory, oxidative stress, synaptoplastic, and neurotransmitter hypotheses are being used to develop new drugs and to study the mechanisms of their action. Important for the understanding of the pathophysiology of AD is the knowledge about the interrelationships, interactions, and synergies of the cellular pathological processes that are considered to be key in the various AD hypotheses.
Key words: Alzheimer´s disease, amyloid beta, biomarker, hypothesis, metabolism, mitochondrial dysfunction, neuroplasticity, neuroinflammation, neurotransmission, tau
ÚVOD
Alzheimerova nemoc (AD) je progredující neurodegenerativní onemocnění provázené ztrátou neuronů, především v mozkové kůře a hipokampu. Poškození nebo ztráta mozkových buněk a narušená neurochemie, neurogeneze a synaptická a nesynaptická neuroplasticita vedou ke kognitivnímu poklesu a AD demenci, která představuje 60 až 80 procent případů diagnostikované demence.1 AD je definována jako klinický syndrom demence potvrzený při pitvě nebo in vivo neuropatologickým pozorováním neuritických plaků složených z amyloidu beta (Aβ) a neurofibrilárních klubek (NFT) složených z párových šroubovicových vláken hyperfosforylovaného proteinu tau (P-tau). Mezi další abnormality v mozcích pacientů s AD patří neurozánět, mitochondriální dysfunkce, porucha autofagie, ztráta cholinergních neuronů, hromadění přechodových kovů, poškození cév a chronická hypoperfuze mozkové tkáně.
Na etiologii AD se podílí především patologie Aβ a tauopatie.2,3 Poškození mozkových buněk a neuroplasticity je způsobeno hlavně účinky volných radikálů (oxidačnim a nitrosačnim stresem) a apoptotickými procesy. Hlavními cíli nových potenciálních léčiv AD jsou procesy související s neurotoxicitou Aβ a P-tau, mitochondriální dysfunkcí, metabolickými poruchami a neurozánětem.
Tento přehled shrnuje hlavní hypotézy AD založené na současném poznání změn v neurochemii a synaptické a strukturální neuroplasticitě vedoucích k neurodegeneraci u AD. Patofyziologie AD je prezentována z hlediska dosavadního poznání primárních příčin a spouštěčů AD vedoucích k neurodegeneraci, za které jsou považovány neurotoxicita oligomerů Aβ a tau, metabolické poruchy, neurozánět a mitochondriální dysfunkce, přičemž určující úlohu mohou mít jejich vzájemné interakce.
Z poznání těchto procesů, interakcí a synergií vyplývají buněčné cíle léků pro AD.
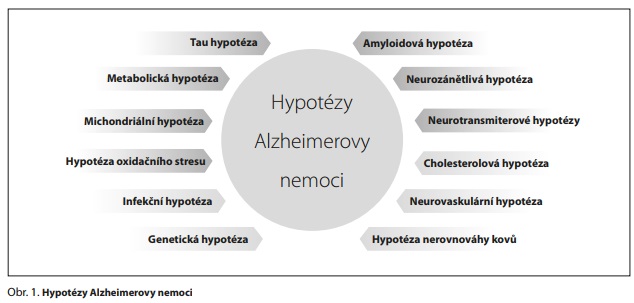
HYPOTÉZY ALZHEIMEROVY NEMOCI
Progresivní neurodegenerace, ztráta synapsí a neuronů a poškození určitých nervových okruhů v mozku vedou ke kognitivní dysfunkci a demenci při AD. Neurodegenerace je ovlivněna věkem, faktory prostředí (stravou, cvičením a životním stylem), kognitivní rezervou, metabolickým a oxidačním stresem, mitochondriální dysfunkcí, poruchou autofagie, genetikou a epigenetikou, cerebrovaskulární dysfunkcí a poruchou hematoencefalické bariéry, neurotoxicitou a neurozánětem.4 Existuje proto mnoho hypotéz AD (obr. 1), které jsou navzájem propojené a odlišují se především tím, co považují za primární příčinu vzniku a rozvoje AD.
Nejpropracovanější a podpořené největším množstvím důkazů jsou amyloidová hypotéza a tau hypotéza. V současné době užívaná schválená léčiva pro AD vycházejí z amyloidové hypotézy a neurotransmiterových hypotéz, především z hypotézy cholinergní a glutamátergní. Masivní metabolické změny v mozkové tkáni podporují metabolickou hypotézu AD. Poruchy metabolismu, a hlavně narušený energetický metabolismus mozku, jsou jednou z prvních změn pozorovaných při rozvoji AD. Vzhledem k tomu, že mitochondrie regulují bioenergetiku, produkci reaktivních forem kyslíku (ROS), apoptózu a kalciovou homeostázu a ovlivňují přímo či nepřímo veškeré nitrobuněčné procesy, je pozornost věnována mitochondriální hypotéze a souvisejícím hypotézám oxidačního stresu a neurozánětu. Podle integrační amyloidové-tau-mitochondriální hypotézy jsou pro rozvoj AD rozhodující interakce rizikových faktorů a biomarkerů a jejich vzájemná synergie spíše než účinky jednoho určitého faktoru.5
Amyloidová hypotéza
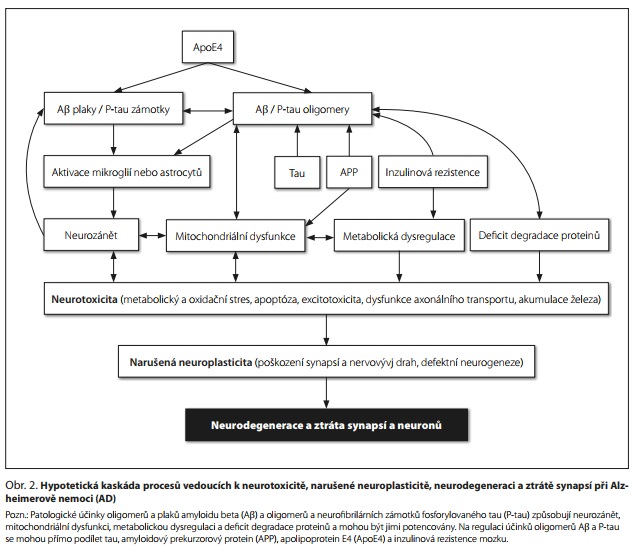
Původní kaskádní amyloidová hypotéza AD předpokládala, že mimobuněčné depozity Aβ (senilní plaky) dávají impuls pro spuštění kaskády patologických procesů vedoucí k tvorbě neurofibrilárních klubek tau proteinu, ztrátě synapsí a mozkových buněk a demenci.6 Za počáteční spouštěče patofyziologie AD u velké části AD s časným nástupem (EOAD) a částečně u sporadické formy AD s pozdním nástupem (LOAD) je dlouhodobě považována zvýšená produkce nebo zhoršená clearance Aβ. Nověji se ukázalo, že za amyloidovou toxicitu u AD jsou odpovědné především rozpustné oligomery Aβ, které narušují synaptickou i nesynaptickou neuroplasticitu.7 Podle hypotézy toxických oligomerů zahrnuje kaskáda neurotoxických účinků Aβ a tau metabolickou dysregulaci, neurozánět, mitochondriální dysfunkci a deficit degradace proteinů, což vede k metabolickému a oxidačnímu stresu, apoptóze, excitotoxicitě, dysfunkci axonového transportu a akumulaci železa. Důsledkem je narušená neuroplasticita, neurogeneze a neurovývoj, poškození synapsí a nervových drah a výsledná neurodegenerace a ztráta neuronů (obr. 2).
Patologie amyloidu beta
Aβ vzniká z APP štěpením katalyzovaným β-sekretázou 1 (BACE1) a β-sekretázou. Agregace Aβ zahrnuje tvorbu monomerů, oligomerů, protofibril nebo paranukleů a fibril, přičemž ukládáním fibril vznikají amyloidové plaky.8 Bylo identifikováno mnoho různých oligomerů s různou neurotoxicitou. Hlavními metabolity jsou Aβ40 a Aβ 42, přičemž Aβ42 je více patogenní a rozpustné oligomery Aβ42 jsou stabilizovány v přítomnosti Aβ 40.
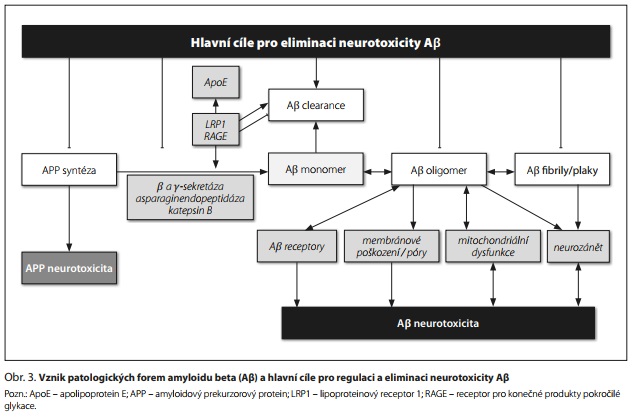
Neurotoxicita Aβ oligomerů je zřejmě spojena s jejich (i) vazbou na Aβ receptory následovanou změnami v clearance Ap nebo v aktivaci různých signálních drah, (ii) vazbou na membránu vedoucí k poškození membrány a tvorbě pórů a (iii) nitrobuněčnou akumulací a narušením buněčných procesů, včetně poškození proteazomu, mitochondriální dysfunkce, poruchy autofagie, neurozánětu, zvýšené produkce ROS a peroxidace lipidů (obr. 3).5
Receptory, které vážou monomerní, oligomerní nebo fibrilární formy Aβ, zahrnují (i) endoteliální receptor pro konečné produkty pokročilé glykace (RAGE) a lipoproteinový receptor 1 (LRP1), které se podílejí na clearance Aβ přes hematoencefalickou bariéru a (ii) řadu postsynaptických receptorů pro Aβ oligomery (např. inzulinový receptor, NGF receptor, glutamátové N-methyl-d-aspartátové (NMDA) receptory a receptory pro kyselinu α-amino-3-hydroxy-5-methyl-4-isoxazolpropionovou (AMPA), glutamátový metabotropní receptor 5, nikotinový acetylcholinový receptor a další).9 Aβ a apolipoprotein E (ApoE) soutěží o vazbu na LRP1 a LRP1 se tak podílí jak na transportu monomerů Aβ, tak na regulaci patogenní role ApoE4.8
Eliminace patologie amyloidu beta
Podle amyloidové hypotézy by farmakologická léčba AD měla být zacílena na eliminaci tvorby a kumulace Aβ v mozku, včetně syntézy APP, clearance Aβ, inhibici agregace AP, a především na potlačení neurotoxických účinků oligomerů a plaků Aβ, jako je neurozánět, mitochondriální dysfunkce, porucha metabolismu acetylcholinu a ovlivnění dalších interakcí a zpětných vazeb mezi neurony, astrocyty, mikrogliemi a vaskulárním systémem. Posílení clearance Aβ přes LRP1 receptory je slibnou terapeutickou strategií pro AD.10 Terapeutickým cílem nových léčiv mohou být samotné oligomery Aβ a jejich tvorba, receptory a navazující signální dráhy, nebo downstream efektory, jako je tau.11
Tau hypotéza
Tau hypotéza AD byla založena na zjištění, že NFT jsou složeny z P-tau. Podle tau hypotézy je primární příčinou (iniciačním faktorem) neurodegenerace u sporadické AD destabilizace mikrotubulů a neurotoxické účinky P-tau a jeho agregátů a tyto účinky tau předchází tvorbu Aβ.12 Oligomerní tau hypotéza předpokládá, že za poškození synapsí při AD jsou odpovědné především tau oligomery, které se tvoří před NFT a způsobují neurodegeneraci a deficit paměti již v časném stadiu AD.13
Fosforylace a defosforylace tau je řízena aktivitami proteinkináz a fosfatáz. Proteinkinázy, které fosforylují tau, zahrnují glykogensyntázukinázu 3 (GSK-3) , cyklin-dependentní kinázu 5 (Cdk5), c-Jun N-terminální kinázu (JNK), kasein kinázu 1, kinázu 1A regulovanou fosforylací tyrosinu s duální specificitou (Dyrk1A), proteinkinázu aktivovanou AMP (AMPK), kinázu 4 regulující afinitu proteinů asociovaných s mikrotubuly k mikrotubulům (MARK4) , proteinkinázu A a tau proteinkinázy. Na defosforylaci tau se mohou podílet různé fosfatázy. GSK-3 fosforyluje tau a současně aktivita GSK-3 přispívá k produkci Aβ. Podle GSK-3 hypotézy AD je nadměrná aktivita GSK-3 zodpovědná za hyperfosforylaci tau, zvýšenou produkci Ap, neurozánětlivé reakce zprostředkované mikrogliemi a poškození paměti.14
Patologie tau
Tau protein je protein asociovaný s mikrotubuly a má klíčovou úlohu při stabilizaci mikrotubulů. V neuronech podporuje růst axonů a dendritů a axonální transport. V dospělém mozku se nachází šest izoforem tau (3R0N, 3R1N, 3R2N, 4R0N, 4R1N a 4R2N). Po hyperfosforylaci z mikrotubulů disociuje, čímž je destabilizuje a narušuje jejich funkci. Neurodegenerativní tauopatie jsou spojeny s hyperfosforylací tau a jeho posttranslační modifikací. V mozcích AD pacientů se hromadí tau v hyperfosforylovaném stavu.15
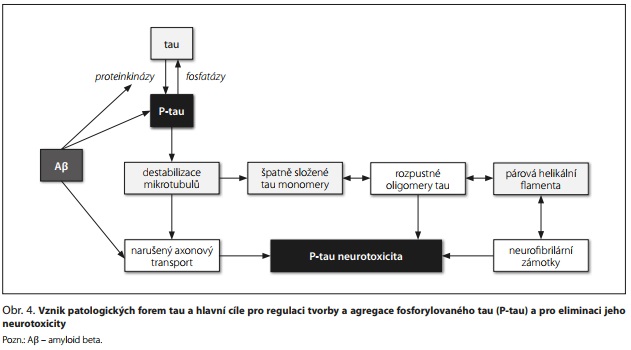
Po uvolnění P-tau z mikrotubulů dochází k jejich destabilizaci a narušení souvisejících buněčných procesů. Uvolněné P-tau se chybně skládají a shlukují, což vede k tvorbě dimerů, trimerů, rozpustných a nerozpustných oligomerů a párových helikálních filament, které nakonec tvoří NFT (obr. 4). Patologické šíření tau umožňují toxické agregáty tau "semen", kdy oligomery nebo fibrily (ale nikoli monomery) tau slouží jako matrice pro chybné skládání nativního tau a mohou se uvolňovat do mimobuněčného prostoru. Mechanismus mezibuněčného šíření tau při neurodegeneraci není zcela objasněn; mohou se na něm podílet exosomy nebo ektosomy, transport heparinsulfátovými proteoglykany, vazba na membránové receptory, endocytóza nebo nanotrubice.16
Patologii tau reguluje oxidační stres, mitochondriální dysfunkce, nerovnováha v aktivitě proteinkináz a fosfatáz a řada proteinových interakcí, především synergie s Aβ, ale také interakce s α-synukleinem, některými proteiny tepelného šoku, imunofiliny, cytoplazmatickými adaptorovými proteiny a peptidylprolylizomerázou.17 Narušený axonový transport a neurotoxicita tau oligomerů a NFT vedou k neuronální dysfunkci, apoptóze a neurodegeneraci.
Eliminace patologie tau
Podle tau hypotézy by farmakologická léčba AD měla být zacílena na eliminaci tvorby, kumulace a agregace P-tau v mozku, stabilizaci mikrotubulů, inhibici mezibuněčného přenosu a uptake P-tau, inhibici proteinkináz a aktivaci fosfatáz, modulaci glykosylace tau a interakce s Aβ (obr. 2 a 4).5
Porucha autofagie
Na patofyziologii AD se podílí také deficit degradace Aβ a tau. S věkem a u neurodegenerativních onemocnění, jako je AD, dochází k narušení eliminace neaktivních nebo toxických proteinů, která je nezbytná pro životaschopnost neuronů. Proteinovou homeostázi udržuje ubikvitin-proteazomový systém (UPS) a lysozomální degradace probíhající mechanismem autofagie. Oba tyto systémy jsou zapojeny do clearance tau.18 Hlavním regulátorem Ap v neuronech je systém UPS, a to buď snížením produkce Aβ, nebo podporou jeho proteolytické degradace. Narušení UPS může vést k abnormální akumulaci Aβ a současně Aβ inhibuje aktivitu UPS.19 U AD byla potvrzena defektní autofagie a mitofagie indukovaná Aβ a tau.20 Systémy UPS a autofagie jsou tedy potenciálními terapeutickými cíli pro AD.
Mitochondriální hypotéza
Mitochondriální kaskádní hypotéza předpokládá, že spuštění a rozvoj AD určuje mitochondriální dysfunkce. Podle této hypotézy mohou primární změny mitochondriální funkce indukovat kaskádu procesů, které vedou k neuropatologickým změnám specifickým pro AD. Interakcí s mitochondriálními proteiny a membránami může být také potencována patologie/neurotoxicita Aβ a tau. Podle původní mitochondriální kaskádní hypotézy je základní úroveň mitochondriální funkce daná geneticky a pokles mitochondriální funkce je dán procesy stárnutí, genetickými faktory a vlivem prostředí. Pokud pokles mitochondriální funkce překročí určitý práh, potom se spustí histologické a patofyziologické změny specifické pro AD, jako jsou změny v APP, sekretovaném APPa, Aβ a tau, neurodegenerace a ztráta synapsí. Amyloidopatie přitom může zpětnovazebně ovlivňovat mitochondriální funkci.21
Podle této hypotézy vzniká neuropatologie AD sekundárně k mitochondriální dysfunkci v okamžiku, kdy s věkem asociovaný pokles mitochondriální funkce dosáhne stavu, kdy nejsou kompenzační mechanismy účinné.22 Předpokládá se, že mitochondriální dysfunkce může vzniknout nezávisle na patologii Aβ a tau a může tuto patologii iniciovat.
Mitochondriální změny, které se mohou podílet na patofyziologii AD, zahrnují narušení mitochondriální dynamiky (směrem k nadměrnému dělení), selhání mitofagie (procesu selektivní degradace mitochondrií autofagií), dysregulaci buněčné energetiky a nitrobuněčného vápníku, oxidační stres a apoptózu.23 Postupně jsou v mitochondriální hypotéze AD přesněji specifikovány mitochondriální změny a jejich souvislost s patologií Aβ a tau.
Mitochondriální hypotéza AD je založena na klíčové roli mitochondrií ve všech buněčných procesech, včetně fyziologického metabolismu, reakce na buněčný stres, buněčné smrti, neuroplastických a neurodegenerativních procesů v mozku regulovaných Aβ, tau, bioenergetikou, apoptózou, neurozánětem a oxidačním stresem.24 Významnou podporou mitochondriální hypotézy je skutečnost, že věk je hlavním rizikovým faktorem pro AD a mitochondriální dysfunkce je charakteristickým znakem stárnutí.
Do mitochondriální hypotézy lze zahrnout dříve formulovanou kalciovou hypotézu AD, podle níž mohou APP a oligomery Aβ zvyšovat vstup Ca2+ do buňky a jeho uvolňování z nitrobuněčných zásob a toto zvýšení nitrobuněčné signalizace Ca2+ způsobuje narušení synaptických procesů odpovědných za učení a paměť a současně dochází k mitochondriální dysfunkci a spuštění apoptózy.25
Poznání úlohy mitochondrií, endoplasmatického retikula (ER) a membránových kontaktních míst mezi mitochondriemi a ER (alternativně označovaných jako membrány asociované s mitochondriemi, MAM) v udržování homeostázy Ca2+, metabolismu fosfolipidů a cholesterolu, importu lipidů do mitochondrií, iniciaci autofagie, mitochondriálním dělení a apoptóze vedlo k MAM hypotéze AD, podle níž je AD především poruchou komunikace mezi ER a mitochondriemi a MAM mohou být cílem nových léků proti AD.26
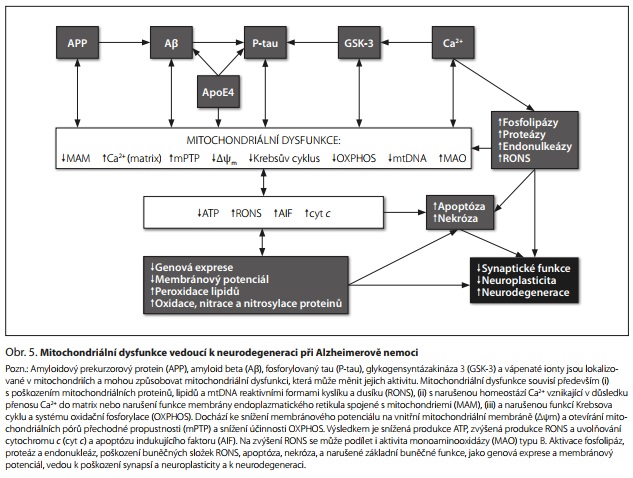
Mitochondriální dysfunkce
Mitochondriální dysfunkce při AD je způsobena a potencována snížením aktivity komplexů dýchacího řetězce a produkce ATP, změnou membránového potenciálu na vnitřní mitochondriální membráně, otevíráním mitochondriálních pórů přechodné propustnosti (mPTP), dysregulací cytosolového kalcia, zvýšenou tvorbou ROS, poškozením mitochondriální DNA (mtDNA), narušením axonového transportu mitochondrií a poruchou mitochondriální dynamiky a biogeneze.5,24,27
Změna aktivity mitochondriálních enzymů při AD je nejlépe prokázána pro snížení aktivity cytochrom c oxidázy (COX, komplex IV) a pro snížení maximální kapacity elektronového transportního systému měřené jako kinetika spotřeby kyslíku mitochondriemi.28 Úlohu mtDNA v patogenezi AD podporují data získaná pomocí AD cybridů (přenosu mtDNA mezi buňkami), které mimo jiné vykazují sníženou aktivitu COX, zvýšený nitrobuněčný Aβ a zvýšenou apoptotickou aktivitu.27 Je akceptováno, že synaptická mitochondriální dysfunkce může být predisponujícím faktorem pro AD, přičemž k rozvoji AD může významně přispět také narušená mitofagie nebo biogeneze.20
Snížená produkce ATP vede k narušení veškerých buněčných procesů, včetně poruch iontových transmembránových gradientů a membránových potenciálů, uvolňování glutamátu a zvýšeného vstupu vápenatých iontů do buňky přes napěťově a ligandem řízené kanály (především přes glutamátové NMDA receptory). Dlouhodobě zvýšená cytosolová koncentrace Ca2+ potencuje účinky Ca2+-aktivovaných fosfolipáz, proteáz a endonukleáz, které nadměrně degradují membrány, proteiny a nukleové kyseliny. Zvýšené nitrobuněčné Ca2+ také způsobuje otevírání mPTP, depolarizaci vnitřní mitochondriální membrány, snížení produkce ATP, nadprodukci ROS, vychytávání antiapoptotických a uvolňování proapoptotických faktorů.29 Proteolytické, oxidační a nitrosační procesy vedou k poškození buňky, včetně mitochondrií samotných. V mozku dochází k poškození synapsí, neuronů a glií a k neurodegeneraci (obr. 5).
Synergie neurotoxicity mitochondriální dysfunkce, amyloidu beta a tau
Potvrzená lokalizace a synergická toxicita APP, Aβ a tau v synaptických mitochondriích ukazují, že mitochondriální dysfunkce při AD může být potencována patologií amyloidu a tau.5,27 Navíc, primární příčinou onemocnění nemusí být pouze mitochondriální dysfunkce, amyloidopatie nebo tauopatie, ale také změny aktivity faktorů, které je mohou vyvolávat a které jsou lokalizovány v mitochondriích, jako je ApoE4, GSK-3 a monoaminooxidáza (MAO). Zdá se, že AD může mít vícero iniciačních patologických faktorů, které se vzájemně ovlivňují.
Na neurodegeneraci při AD se podílí synaptotoxicita a neurotoxicita spojená jak s mitochondriální dysfunkcí, tak s patologií amyloidových a tau oligomerů a agregátů. APP způsobuje narušení schopnosti mitochondrií pufrovat Ca2+, zhoršení mitochondriální dynamiky, snížení exprese peroxisomového PGC-1α (klíčový regulátor energetického metabolismu) a narušení mitochondriální biogeneze a mitofagie. Účinky Aβ jsou spojeny s narušením cytoplazmatické membrány, inhibicí importu proteinů do mitochondrií a inhibicí mitochondriálních enzymů včetně respiračních komplexů, 17β-hydroxysteroiddehydrogenázy typu 10 (HSD10), oxoglutarátdehydrogenázového komplexu (OGDC) a dalších. Mitochondriální cíle A0 zahrnují presekvenční proteázu (PreP), metaloproteázu odpovědnou za degradaci AP v matrix a další enzymy degradující A0, adenin nukleotid translokázu (ANT1) a napěťově řízený aniontový kanál 1 (VDAC1).5
Patologie P-tau u AD je spojena s narušením mitochondriálního transportu, regulací mitochondriální dynamiky a ovlivněním mitochondriální bioenergetiky. P-tau zvyšuje oxidační stres a snižuje expresi komplexů I a V, aktivitu komplexu I, produkci ATP, pufrační kapacitu mitochondrií pro Ca2+ a membránový potenciál. P-tau může zvýšit expresi mitochondriální peptidyl-prolyl cis-trans izomerázy (PPIF) a způsobit otevírání mPTP, zvýšit nitrosylaci proteinu podobného dynaminu-1 (Drp1) a interakce s ANT1 a VDAC1.30
Regulace mitochondriální dysfunkce
Vzhledem k provázanosti buněčných procesů regulovaných mitochondriemi existuje řada zpětnovazebných procesů, které mitochondriální dysfunkci zesilují a které jsou potenciálními cíli nových léčiv. Terapie AD založená na regulaci mitochondriální dysfunkce (obr. 5) je zacílena především na snížení agregace Aβ a hyperfosforylace tau, snížení produkce ROS, aktivaci mitofagie a mitochondriální biogeneze, deaktivaci neurozánětu, snížení dělení mitochondrií, změny aktivit řady mitochondriálních proteinů a mitochondriálních i jaderných transkripčních faktorů, transport Ca2+ do matrix, import proteinů do mitochondrií translokázami vnější a vnitřní membrány a snížení akumulace cholesterolu v mitochondriích.31 Mitochondriální dysfunkce a neurodegenerace vyvolané Aβ nebo P-tau mohou být sníženy aktivací PGC-1α agonisty jaderného receptoru PPARy, jako je metformin, resveratrol a hopeahainol A. Protože zásah do mitochondriální funkce ovlivní prakticky všechny buněčné funkce, je vhodné se zaměřit při vývoji a testování nových léků pro AD na interakce mitochondriálních proteinů s Aβ a tau a současně testovat mitochondriální toxicitu těchto látek.
Hypotéza oxidačního stresu
Podle hypotézy oxidačního stresu je příčinou rozvoje AD oxidační stres, kdy poškození mozkových buněk ROS přispívá k neurodegeneraci a poklesu kognitivních funkcí. Na zvýšené produkci reaktivních forem kyslíku a dusíku (RONS) se přitom mohou podílet Aβ a P-tau.32,33
Zvýšený oxidační stres v mozku osob s AD a jeho podíl na neurodegeneraci je doložen pozorováním snížené antioxidační ochrany, zvýšené koncentrace neurotoxických stopových prvků (železa, hliníku, rtuti) schopných stimulovat tvorbu ROS, zvýšené peroxidace lipidů a oxidace proteinů a nukleových kyselin, sníženého energetického metabolismu a zvýšené produkce ROS mitochondriemi, zvýšených konečných produktů pokročilé glykace a dalších. ROS mohou spouštět zánětlivou odezvu a naopak, zánět indukuje oxidační stres. Není jasné, zda je zvýšený oxidační stres primární nebo sekundární v kaskádě procesů vedoucích k neurodegeneraci při AD; v počáteční fázi vývoje AD může být patologie Aβ a P-tau důsledkem oxidačního stresu.
Antioxidační terapie
Z hypotézy oxidačního stresu vyplývá antioxidační strategie při léčbě AD. Interpretace hypotézy, že antioxidanty jsou jednoznačně prospěšné v léčbě AD, je zřejmě příliš zjednodušená, neboť nebere v úvahu fyziologickou úlohu ROS. Při farmakologické regulaci oxidačního stresu je nutné mít na vědomí, že oxidační stres může vyvolat kompenzační reakce a následné adaptace, které mohou být protektivní proti oxidačnímu poškození mozkových buněk.34
Synaptoplastická hypotéza
Synaptoplastická hypotéza AD považuje za hlavní příčinnou událost ve vývoji onemocnění spojení přirozené neurodegenerace a maladaptivní synaptoplastické odpovědi pro specifickou subpopulaci mozkových buněk, v nichž potom dochází k iniciaci amyloidové a tau patologie.35 Synaptoplastická hypotéza AD předpokládá, že poškození neuroplasticity (věkem nebo geneticky a procesy podmíněnými vnějším prostředím) vyvolává nejprve adaptivní upregulaci buněčné aktivity, včetně nadměrné fosforylace tau a obratu APP, což následně vede ke tvorbě NFT a amyloidových plaků a nakonec ke ztrátě neuronů, dendritů a synapsí. Tato hypotéza sjednocuje amyloidovou a tau hypotézu AD tím, že předpokládá, že jak amyloidová, tak tau patologie jsou nezávislými projevy poškození neuroplasticity jakožto základního procesu, jímž mohou různé genotypy AD spouštět identický klinický a neuropatologický fenotyp.36
Nověji se předpokládá, že časná kognitivní porucha u AD je z velké části výsledkem synaptické dysfunkce způsobené oligomery Aβ. Oligomery Aβ mohou narušit synaptickou plasticitu vazbou na membrány a modulací membránových procesů. Výsledkem může být poškození synapsí a neuronů v důsledku (i) stimulace gliových buněk a uvolňování zánětlivých cytokinů nebo (ii) zvýšení mimobuněčné koncentrace glutamátu a zvýšení aktivace NMDA receptorů.37
Narušená neuroplasticita
Neuroplasticita, jakožto schopnost mozku přizpůsobovat se a reorganizovat své struktury a funkce v reakci na vnější i vnitřní podněty, je klíčový koncept zahrnutý ve všech biologických hypotézách neuropsychiatrických a neurodegenerativních onemocnění, včetně AD. Pojem neuroplasticita zahrnuje synaptickou plasticitu i nesynaptickou (strukturální) plasticitu.38 Spojení neuroplasticity s patologií AD může být realizováno přes vznik dysfunkčních synapsí a spojů nebo přes narušení funkce nervových drah v mozku. Kognitivní rezerva, která se podílí na odolnosti vůči kognitivnímu poklesu při AD, je zřejmě určena do značné míry individuální pozitivní neuroplasticitou.39
Velikost a dynamika neuroplastických změn je určena věkem a genetickými a environmentálními faktory. Změny synaptické plasticity v hippocampu, amygdale, striatu a mozkové kůře mají určující úlohu v procesech učení a paměti, které jsou při AD narušeny.40 Nalezení biomarkerů, které zachycují časné změny v konektivitě a neuroplasticitě při nástupu AD, může pomoci v hledání nových léčiv účinných v časných fázích onemocnění.
Neurozánětlivá hypotéza
Neurozánětlivá hypotéza AD pokládá za určující proces v rozvoji onemocnění chronický zánět v mozku, který může způsobit poškození synapsí a neuronů a postupné zhoršování kognitivních funkcí. Tato hypotéza je založena na pozorování, že nástup a progrese AD jsou provázeny aktivací mikroglií a astrocytů a že dlouhodobý zánětlivý proces v mozku vede k oxidačnímu a nitrosačnímu stresu, poškození neuroplasticity a k neurodegeneraci.41
Produkce zánětlivých mediátorů mikrogliemi může být aktivována agregáty Aβ a P-tau a může také souviset s mitochondriální dysfunkcí, změnami neurální architektury a neurogeneze, sníženou clearance Aβ a zvýšenou fosforylací tau kinázami regulovanými interleukiny, jako jsou Cdk5, GSK-3 a mitogenem aktivované proteinkinázy (MAPK). Dále může docházet ke zvýšenému vstupu zánětlivých mediátorů z periferie přes porušenou hematoencefalickou bariéru.
Klíčovou úlohu má v neurozánětlivé dráze u AD in-flamasom NLRP3. Oligomery Aβ a agregáty tau mohou stimulovat sestavení inflamasomu NLRP3 v mikrogliích a astrocytech s následnou aktivací kaspázy 1 a uvolněním interleukinů; spustí se tak patofyziologické změny vedoucí k neurodegeneraci a kognitivnímu poklesu při AD.42 Neurozánět může zahrnovat i exosomy, které transportují Aβ, tau, zánětlivé faktory a další patogenní látky mezi mikrogliemi, astrocyty a neurony, přičemž aktivace inflamasomu může regulovat uvolňování exosomů a naopak.43
Regulace neurozánětu
Vzhledem k roli neurozánětu v patofyziologii AD se nabízí léčba protizánětlivými léky. Nejčastěji používanými protizánětlivými léky jsou nesteroidní antiflogistika (NSAID), která inhibují cyklooxygenázy. Účinnost NSAID a paracetamolu na progresi kognitivního poklesu při mírné kognitivní poruše (MCI) a AD však nebyla potvrzena. Pouze užívání diklofenaku bylo spojeno s pomalejším kognitivním poklesem u AD.44 Patologie Aβ a tau a inzulinová rezistence jsou regulovány cytokiny, jako je faktor nádorové nekrózy (TNF). Inhibice TNF proto může být vhodným terapeutickým cílem pro nové léky proti AD. Možnosti modifikace progrese AD založené na neurozánětlivé hypotéze zahrnují tyto přístupy: (i) zásah do integrity mozkového lymfatického systému, (ii) interakce mezi neurony, mikrogliemi a astrocyty, (iii) stárnutí mozkových buněk a (iv) osa mozek-střevo.45
Metabolická hypotéza
Podle metabolické hypotézy je sporadická AD s pozdním nástupem způsobena snížením metabolismu glukózy v mozku a souvisejícími důsledky v důsledku inzulinové rezistence a narušené inzulinové signalizace v mozku.46,47 Metabolická hypotéza považuje za klíčové v nástupu a progresi AD narušení energetického metabolismu a metabolismu glukózy v mozku, které přispívají k poškození synapsí, neuronů, neurodegeneraci a symptomům AD. Narušená regulace metabolismu glukózy při AD může být spojena patologií Aβ a P-tau a také s inzulinovou rezistencí v mozku a může vést k mitochondriální dysfunkci, zvýšenému oxidačnímu stresu a neurozánětu.
K pojetí AD jako metabolického onemocnění přispívají účinky ApoE4, který kromě úlohy v lipidovém metabolismu se podílí také na neurodegeneraci přes Aβ a NFT, neurozánětu, oxidačním stresu a poruše signalizace inzulinu v mozku.
Narušený metabolismus
Metabolická hypotéza vychází z pozorování, že neurodegenerace a pokles kognitivních funkcí při AD jsou spojeny s narušeným metabolismem jak na systémové, tak na nitrobuněčné mitochondriální úrovni. Poruchy energetického metabolismu a snížené využití glukózy mohou být spojeny s narušenou inzulinovou signalizací v mozku, tj. s abnormalitami v signálních cestách inzulinu a inzulinu podobných růstových faktorů.48 K rozvoji AD může přispět (i) hypertenze přes vaskulární změny vedoucí k hypoperfúzi, ischémii a hypoxii mozku; (ii) hyperlipidémie přes narušení hematoencefalické bariéry, zvýšené ukládání Aβ, hyperfosforylaci tau a neurozánět; (iii) obezita ve středním věku přes inzulinovou rezistenci; a (iv) diabetes mellitus 2. typu (T2DM) přes inzulinovou rezistenci a narušenou funkci inzulinového receptoru, toxicitu vyvolanou hyperglykémií, konečné produkty pokročilé glykace, neurozánět, cerebrovaskulární poškození a další.49 Inzulinová rezistence nemusí být přímou primární příčinou AD, ale zvýšení neurozánětu, mitochondriální dysfunkce, zvýšení produkce ROS a zvýšení tvorby a ukládání Aβ a P-tau související s inzulinovou rezistencí mohou zvýšit riziko vzniku AD nebo urychlit progresi onemocnění.
Inzulin v mozku se přes aktivaci svého receptoru podílí na kontrole příjmu potravy a na regulaci kognitivních funkcí. Vlivem vnějších faktorů se může vyvinout inzulinová rezistence v mozku a ta může vést ke snížení kognitivních funkcí. Inzulinová rezistence může přispívat k neurodegeneraci související s tau u preklinické AD, ale nebylo dosud prokázáno propojení inzulinové rezistence v mozku se specifickou patologií A0 a tau měřenou pomocí validovaných biomarkerů AD (Aβ42, P-Tau181 a celkový tau). Předpokládá se, že zranitelnost různých oblastí mozku vůči patologii Aβ a tau souvisí s lokální expresí glukózových transportérů a inzulinových signalizačních genů; oligomery Aβ zřejmě vedou k inzulinové rezistenci v mozku a ta podporuje hyperfosforylaci tau.50 Nitrobuněčné signální dráhy inzulinu a inzulinu podobného růstového faktoru zahrnují inhibici GSK-3 a aktivaci proteinkinázy Akt a komplexu 1 savčího cíle rapamycinu (mTORC1), což podporuje syntézu lipidů a proteinů a metabolismus a biogenezi mitochondrií prostřednictvím dráhy od PGC-1β k jaderným transkripčním faktorům (NRF) 1 a 2. Podílejí se tak na regulaci buněčné proliferace, migrace, glukózy, apoptózy a neuroplasticity; mimo jiné dochází také k regulaci fosforylace tau.
Regulace metabolických procesů
Vzhledem k provázanosti AD a metabolických poruch lze terapeutickou strategii zaměřit jak na hledání nových látek pro regulaci mozkového metabolismu, tak na testování účinnosti léčiv již schválených pro léčbu metabolických onemocnění spojených s AD, jako je T2DM. Pomocí pokročilých spektroskopických metod a metabolomiky se současně hledají nové metabolické markery a další buněčné metabolické procesy, které jsou spojeny s rozvojem AD a které mohou být potenciálními cíli nových léčiv.
Neurotransmiterové hypotézy
Při AD dochází ke změnám v acetylcholinovém systému a v dalších neurotransmiterových systémech, především glutamátovém, dopaminovém, noradrenalinovém, serotoninovém a histaminovém.51
Cholinergní hypotéza
Podle cholinergní hypotézy AD je zhoršení kognitivních funkcí osob s AD do značné míry způsobeno progresivní ztrátou cholinergní inervace v mozkové kůře spojenou s neurodegenerací cholinergních neuronů v předním mozku a související ztrátou cholinergní neurotransmise v mozkové kůře a dalších mozkových oblastech.52 Původní cholinergní hypotéza AD byla založena (i) na pozorování ztráty centrálních cholinergních neuronů a na zjištění, že nucleus basalis Meynerti, který je hlavním zdrojem cholinergní inervace mozku, včetně neokortexu, podléhá degeneraci při AD,53 a (ii) na účincích anticholinergních látek (skopolaminu) a reverzibilních inhibitorů acetylcholinesterázy (AChE) (fyzostigminu) na zhoršení či zlepšení paměti.
Změny v neurotransmisi, které byly podkladem pro revidovanou cholinergní hypotézu, zahrnují sníženou cholinergní inervaci mozkové kůry, sníženou kortiko-kortikální glutamátovou neurotransmisi, sníženou senzitivitu muskarinových acetylcholinových M1 receptorů a sníženou produkci glutamátu. Hypotéza předpokládá, že tyto změny, spolu s amyloidovou a tau patologií, vedou ke vzniku klinických příznaků AD.54
Podkladem pro cholinergní hypotézu AD byl pozorovaný cholinergní deficit, tj. snížená koncentrace acetylcholinu a aktivity cholinacetyltransferázy (ChAT), ztráta cholinergních neuronů a účinky inhibitorů AChE. Také byl pozorován vliv oligomerů A0 na acetylcholinové synapse. Skutečnost, že ztráta cholinergních neuronů přispívá k poruchám paměti a pozornosti, vede k očekávání, že léky potencující centrální cholinergní systém mohou být slibné v léčbě pacientů s AD.55
Cholinergní hypotéza byla podpořena terapeutickými účinky inhibitorů cholinesteráz (takrinu, donepezilu, rivastigminu, galantaminu a dalších). Terapie inhibitory cholinesteráz je schválena jako symptomatická léčba AD, i když nelze vyloučit, že dlouhodobé podávání těchto léků může mít také chorobu modifikující účinky vedoucí ke zpomalení neurodegenerace mozku, neboť inhibitory cholinesteráz mohou aktivovat dráhu přežití neuronů PI3K/Akt/mTOR (fosfoinositid 3-kináza/proteinkináza B/savčí cíl rapamycinu), což vede k inhibici GSK-3 a snížení hyperfosforylace tau a tvorby NFT, inhibici proapoptotických faktorů, inhibici autofagie a zvýšení antioxidační ochrany.56
Glutamátergní hypotéza
Glutamátergní hypotéza AD předpokládá, že progresivní kognitivní pokles AD je způsoben ztrátou neuronů v důsledku nadměrné aktivace extrasynaptických glutamátových NMDA receptorů (např. v důsledku abnormální funkce přenašeče excitačních aminokyselin nebo sníženého vychytávání glutamátu ze synaptické štěrbiny gliovými buňkami) a následného zvýšení nitrobuněčného Ca 2+. Excitační glutamátová neurotransmise přes NMDA receptor je určující pro synaptickou plasticitu, přežití neuronů, učení a paměť. Nadměrná aktivita NMDA receptorů ale způsobuje nadměrný vstup Ca2+ do buňky a způsobuje tak oxidační stres a excitotoxicitu, která vedou k neurodegeneraci u AD.57 Dysfunkce glutamátových receptorů a přenašečů a homeostáze nitrobuněčného vápníku může být spojena s patologií Aβ a P-tau. 58
Zapojení dysfunkce glutamátové neurotransmise, resp. glutamátové excitotoxicity, do etiologie AD je podpořeno terapeutickými účinky memantinu, který je nekompetitivním antagonistou glutamátových NMDA receptorů. Pozornost je věnována propojení cholinergní a glutamátergní neurotransmise a možnostem její regulace při AD.51
ZÁVĚR
Pokrok v poznání patofyziologie AD je založen na rozpoznání primárních příčin onemocnění, které lze odvodit z rizikových faktorů a biomarkerů měřitelných v časném stadiu onemocnění. Vhled do rizikových faktorů, biomarkerů, časového průběhu abnormalit biomarkerů, neurotoxicity Aβ a tau, metabolické a mitochondriální dysregulace, oxidačního stresu, neurozánětu a narušení signálních drah a neuroplasticity, spolu s novými poznatky o mechanismech neurodegenerace, podporují propojení především amyloidové, tau, mitochondriální, metabolické, neurozánětlivé a synaptoplastické hypotézy. Integrační amyloidová-tau-mitochondriální hypotéza předpokládá, že různé rizikové faktory a některé biomarkery a metabolické změny mohou spustit rozvoj AD a neurodegeneraci na základě vzájemných vazeb a synergických interakcí především mezi neurotoxickými účinky Aβ oligomerů, tau oligomerů a mitochondriální dysfunkce.5 Pro pochopení patofyziologie AD jsou určující jednak nové poznatky o mechanismech neurodegenerace a její regulace, jednak poznatky o vzájemném propojení, interakcích a synergiích buněčných procesů specifikovaných v různých hypotézách AD. Jako nejperspektivnější pro kauzální léčbu AD se jeví zacílení na patologii Aβ a tau a její propojení s mitochondriální dysfunkcí, neurozánětem, oxidačním stresem, metabolickými poruchami, a neurotransmiterovými systémy.
Použité zkratky
Aβ - amyloid beta; AChE - acetylcholinesteráza; AD - Alzheimerova nemoc; AIF - apoptózu indukující faktor; AMPK - proteinkináza aktivovaná AMP; ANT1 - adenin nukleotid translokáza; ApoE - apolipoprotein E; APP - amyloidový prekurzovoý protein; BACE - beta sekretáza; Cdk5 - cyklin-dependentní kináza 5; cyt c - cytochrom c; Drp1 - protein podobný dynaminu-1; Dyrk1A - kináza 1A regulovaná fosforylací tyrosinu s duální specificitou; EOAD - AD s časným nástupem; GSK-3 - glykogensyntázakináza 3; HSD10 - 17β?hydroxysteroiddehydrogenáza typu 10; ChAT - cholinacetyltransferáza; JNK - c-Jun N-terminální kináza; LOAD - sporadická forma AD s pozdním nástupem; LRP1 - lipoproteinový receptor 1; MAM - membrány endoplazmatického retikula asociované s mitochondriemi; MAO - monoaminooxidáza; MAPK - mitogenem aktivovaná proteinkináza; MARK4 - kináza 4 regulující afinitu proteinů asociovaných s mikrotubuly k mikrotubulům; mtDNA - mitochondriální DNA; MCI - mírná kognitivní porucha; mPTP - mitochondriální pór přechodné propustnosti; mTOR - savčí cíl rapamycinu; NFT - neurofibrilární klubko; NMDA - N-methyl-d-aspartát; NRF - jaderný respirační faktor; NSAID - nesteroidní antiflogistika; OGDC - oxoglutarát-dehydrogenázový komplex; OXPHOS - oxidační fosforylace; PGC-1α - peroxisomovým proliferátorem aktivovaný receptor gama koaktivátor 1-alfa; PPIF - mitochondriální peptidyl-prolyl cis-trans izomeráza; PreP - presekvenční proteáza; P-tau - fosforylovaný tau; RAGE - receptor pro konečné produkty pokročilé glykace; RONS - reaktivními formy kyslíku a dusíku; ROS - reaktivní formy kyslíku; T2DM - diabetes mellitus 2. typu; TNF - faktor nádorové nekrózy; VDAC1 - napěťově řízený aniontový kanál.
LITERATURA
- 1. 2023 Alzheimer´s disease facts and figures. Alzheimers Dement 2023; 19 (4): 1598-1695.
- 2. Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer´s disease. Alzheimers Dement 2018; 14 (4): 535-562.
- 3. McKhann GM, Knopman DS, Chert-kow H, Hyman BT, Jack CR Jr., Kawas CH et al. The diagnosis of dementia due to Alzheimer´s disease: recommendations from the National Institute on Aging-Alzheimer´s Association workgroups on diagnostic guidelines for Alzheimer´s disease. Alzheimers Dement 2011; 7 (3): 263-269.
- 4. Wareham LK, Liddelow SA, Temple S, Benowitz LI, Di Polo A, Wellington C et al. Solving neurodegeneration: common mechanisms and strategies for new treatments. Mol Neurodegener 2022; 17 (1): 23.
- 5. Fišar Z. Linking the amyloid, tau, and mitochondrial hypotheses of Alzheimer´s disease and identifying promising drug targets. Biomolecules 2022; 12 (1676): 1-43.
- 6. Hardy JA, Higgins GA. Alzheimer´s disease: the amyloid cascade hypothesis. Science 1992; 256 (5054): 184-185.
- 7. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer´s disease at 25 years. EMBO Mol Med 2016; 8 (6): 595608.
- 8. Hampel H, Hu Y, Hardy J, Blennow K, Chen C, Perry G et al. The amyloid-beta pathway in Alzheimer´s disease: a plain language summary. Neurodegener Dis Manag 2023; 13 (3): 141149.
- 9. Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM. Amyloid-beta Receptors: The Good, the Bad, and the Prion Protein. J Biol Chem 2016; 291 (7) : 3174-3183.
- 10. Shinohara M, Tachibana M, Kanekiyo T, Bu G. Role of LRP1 in the pathogenesis of Alzheimer´s disease: evidence from clinical and preclinical studies. J Lipid Res 2017; 58 (7): 1267-1281.
- 11. Overk CR, Masliah E. Toward a unified therapeutics approach targeting putative amyloid-beta oligomer receptors. Proc Natl Acad Sci U S A 2014; 111 (38): 13680-13681.
- 12. Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer´s disease. Alzheimers Dement 2021; 17 (1): 115-124.
- 13. Cardenas-Aguayo Mdel C, Gomez-Virgilio L, DeRosa S, Meraz-Rios MA. The role of tau oligomers in the onset of Alzheimer´s disease neuropathology. ACS Chem Neurosci 2014; 5 (12): 1178-1191.
- 14. Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer´s disease. J Neurochem 2008; 104 (6): 14331439.
- 15. Goedert M, Klug A, Crowther RA. Tau protein, the paired helical filament and Alzheimer´s disease. J Alzheimers Dis 2006; 9 (3 Suppl.): 195-207.
- 16. Zhang H, Cao Y, Ma L, Wei Y, Li H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer´s Disease. Front Cell Dev Biol 2021; 9: 707268.
- 17. Muralidar S, Ambi SV, Sekaran S, Thirumalai D, Palaniappan B. Role of tau protein in Alzheimer´s disease: The prime pathological player. Int J Biol Macromol 2020; 163: 1599-1617.
- 18. Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 2013; 105: 49-59.
- 19. Hong L, Huang HC, Jiang ZF. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer´s disease. Neurol Res 2014; 36 (3): 276-282.
- 20. Reddy PH, Oliver DM. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer´s Disease. Cells 2019; 8 (5).
- 21. Swerdlow RH, Khan SM. A "mitochondrial cascade hypothesis" for sporadic Alzheimer´s disease. Med Hypotheses 2004; 63 (1): 8-20.
- 22. Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer´s disease. J Alzheimers Dis 2018; 62 (3): 1403-1416.
- 23. Ashleigh T, Swerdlow RH, Beal MF. The role of mitochondrial dysfunction in Alzheimer´s disease pathogenesis. Alzheimers Dement 2023; 19 (1): 333-342.
- 24. Hroudová J, Singh N, Fišar Z. Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer´s disease. Biomed Res Int 2014; 2014: 175062.
- 25. Berridge MJ. Calcium hypothesis of Alzheimer´s disease. Pflugers Arch 2010; 459 (3): 441-449.
- 26. Schon EA, Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 2013; 55: 26-36.
- 27. Weidling IW, Swerdlow RH. Mitochondria in Alzheimer´s disease and their potential role in Alzheimer´s proteostasis. Exp Neurol 2020; 330: 113321.
- 28. Fišar Z, Hansíková H, Křížová J, Jirák R, Kitzlerová E, Zvěřová M et al. Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer´s disease and depressive disorder. Mitochondrion 2019; 48: 67-77.
- 29. Hroudová J, Fišar Z. Connectivity between mitochondrial functions and psychiatric disorders. Psychiatry Clin Neurosci 2011; 65 (2): 130-141.
- 30. Perez MJ, Jara C, Quintanilla RA. Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front Neurosci 2018; 12: 441.
- 31. Mi Y, Qi G, Brinton RD, Yin F. Mitochondria-targeted therapeutics for Alzheimer´s disease: the good, the bad, the potential. Antioxid Redox Signal 2021; 34 (8): 611-630.
- 32. Padurariu M, Ciobica A, Lefter R, Serban IL, Stefanescu C, Chirita R. The oxidative stress hypothesis in Alzheimer´s disease. Psychiatr Danub 2013; 25 (4): 401-409.
- 33. Markesbery WR. Oxidative stress hypothesis in Alzheimer´s disease. Free Radic Biol Med 1997; 23 (1): 134-147.
- 34. Pratico D. Oxidative stress hypothesis in Alzheimer´s disease: a reappraisal. Trends Pharmacol Sci 2008; 29 (12): 609-615.
- 35. Neill D. Alzheimer´s disease: maladaptive synaptoplasticity hypothesis. Neurodegeneration 1995; 4 (2): 217-232.
- 36. Mesulam MM. A plasticity-based theory of the pathogenesis of Alzheimer´s disease. Ann N Y Acad Sci 2000; 924: 42-52.
- 37. Li S, Selkoe DJ. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer´s brain. J Neurochem 2020; 154 (6): 583-597.
- 38. Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008; 33 (1): 18-41.
- 39. Vance DE, Roberson AJ, McGuinness TM, Fazeli PL. How neuroplasticity and cognitive reserve protect cognitive functioning. J Psychosoc Nurs Ment Health Serv 2010; 48 (4): 23-30.
- 40. Merceron-Martinez D, Ibaceta-Gonzalez C, Salazar C, Almaguer-Melian W, Bergado-Rosado JA, Palacios AG. Alzheimer´s disease, neural plasticity, and functional recovery. J Alzheimers Dis 2021; 82 (S1): S37-S50.
- 41. Long HZ, Zhou ZW, Cheng Y, Luo HY, Li FJ, Xu SG et al. The role of microglia in Alzheimer´s disease from the perspective of immune inflammation and iron metabolism. Front Aging Neurosci 2022; 14: 888989.
- 42. Pereira CF, Santos AE, Moreira PI, Pereira AC, Sousa FJ, Cardoso SM et al. Is Alzheimer´s disease an inflammasomopathy? Ageing Res Rev 2019; 56: 100966.
- 43. Weng S, Lai QL, Wang J, Zhuang L, Cheng L, Mo Y et al. The role of exosomes as mediators of neuroinflammation in the pathogenesis and treatment of Alzheimer´s disease. Front Aging Neurosci 2022; 14: 899944.
- 44. Rivers-Auty J, Mather AE, Peters R, Lawrence CB, Brough D. Anti-inflammatories in Alzheimer´s disease-potential therapy or spurious correlate? Brain Commun 2020; 2 (2): fcaa109.
- 45. Wong-Guerra M, Calfio C, Maccioni RB, Rojo LE. Revisiting the neuroinflammation hypothesis in Alzheimer´s disease: a focus on the druggability of current targets. Front Pharmacol 2023; 14: 1161850.
- 46. Hoyer S. Is sporadic Alzheimer disease the brain type of non-insulin dependent diabetes mellitus? A challenging hypothesis. J Neural Transm (Vienna) 1998; 105 (4-5): 415-422.
- 47. Morgen K, Frolich L. The metabolism hypothesis of Alzheimer´s disease: from the concept of central insulin resistance and associated consequences to insulin therapy. J Neural Transm (Vienna) 2015; 122 (4): 499-504.
- 48. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer´s disease - is this type 3 diabetes? J Alzheimers Dis 2005; 7 (1): 63-80.
- 49. Ezkurdia A, Ramirez MJ, Solas M. Metabolic syndrome as a risk factor for Alzheimer´s disease: a focus on insulin resistance. Int J Mol Sci 2023; 24 (5) : 4354.
- 50. Mullins RJ, Diehl TC, Chia CW, Kapogiannis D. Insulin resistance as a link between amyloid-beta and tau pathologies in Alzheimer´s disease. Front Aging Neurosci 2017; 9: 118.
- 51. Francis PT. The interplay of neurotransmitters in Alzheimer´s disease. CNS Spectr 2005; 10 (11 Suppl. 18): 6-9.
- 52. Hampel H, Mesulam MM, Cuello AC, Farlow MR, Giacobini E, Grossberg GT et al. The cholinergic system in the pathophysiology and treatment of Alzheimer´s disease. Brain 2018; 141 (7): 1917-1933.
- 53. Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol 1981; 10 (2): 122-126.
- 54. Francis PT, Sims NR, Procter AW, Bowen DM. Cortical pyramidal neurone loss may cause glutamatergic hypoactivity and cognitive impairment in Alzheimer´s disease: investigative and therapeutic perspectives. J Neuro-chem 1993; 60 (5) : 1589-1604.
- 55. Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM. Alzheimer´s disease: Targeting the cholinergic system. Curr Neuropharmacol 2016; 14 (1): 101115.
- 56. Moreira N, Lima J, Marchiori MF, Carvalho I, Sakamoto-Hojo ET. Neuroprotective effects of cholinesterase inhibitors: current scenario in therapies for Alzheimer´s disease and future perspectives. J Alzheimers Dis Rep 2022; 6 (1): 177-193.
- 57. Wang R, Reddy PH. Role of Glutamate and NMDA Receptors in Alzheimer´s Disease. J Alzheimers Dis 2017; 57 (4): 1041-1048.
- 58. Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid b peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci 2013; 38 (1): 6-23.