Česká a slovenská psychiatrie

Časopis
Psychiatrické společnosti ČLS JEP
a Psychiatrickej spoločnosti SLS
souborný článek / review article
- AKTUÁLNÍ ČÍSLO
- ARCHIV
- VYHLEDÁVÁNÍ
- PERIODIKUM
- REDAKČNÍ RADA
- PŘEDPLATNÉ
- INZERCE
- AKTUALIZOVANÉ
POKYNY
PRO AUTORY
- ISSN 1212-0383




- © Česká a Slovenská psychiatrie 2025
- © Galén 2025
GENETIKA NEUROVÝVOJOVÝCH PORUCH
GENETICS OF NEURODEVELOPMENTAL DISORDERS
Zdeněk Sedláček
Ústav biologie a lékařské genetiky 2. LF UK a FN Motol, Praha
Podpořeno grantem AZV MZ ČR 17-29423A.
SOUHRN
Sedláček Z. Genetika neurovývojových poruch
Neurovývojové poruchy zahrnují širokou škálu fenotypů podmíněných defekty ve vývoji a fungování centrálního nervového systému. Při jejich rozvoji hrají důležitou roli genetické faktory. Náhled na způsob genetické determinace neurovývojových poruch se za posledních několik desetiletí několikrát změnil. Dnes převládá kombinovaný model, podle něhož se na rozvoji těchto poruch podílejí jak běžné genetické varianty s malým účinkem, tak vzácné varianty se silným účinkem, a oba tyto mechanismy působí u každého pacienta vedle sebe. Odhalování polygenních sítí běžných variant je obtížné a první asociované lokusy byly popsány teprve nedávno. Identifikace variant se silným účinkem je jednodušší a byly nalezeny již v několika stovkách genů. Zajímavé je, že tyto geny často zajišťují základní buněčné procesy, a zdá se, že jejich mutace jsou ve většině systémů tolerovány, s výjimkou centrálního nervového systému. Pokrok v identifikaci kauzálních genů umožňuje definovat na základě genetiky nové syndromy zahrnující neurovývojové poruchy. Dnes se to však daří pouze u vysoce penetrantních mutací v genech podmiňujících častější, těžší a fenotypově méně variabilní monogenní syndromy, a proto zůstává mnoho zúčastněných genů a na ně vázaných klinických jednotek stále neodhaleno. Lepší pochopení genetických mechanismů se již dnes odráží v efektivnější genetické diagnostice, a v budoucnu snad umožní i cílenou terapii některých syndromů spojených s neurový-vojovými poruchami.
Klíčová slova: běžná varianta, genetická heterogenita, mentální retardace
SUMMARY
Sedláček Z. Genetics of neurodevelopmental disorders
Neurodevelopmental disorders include a wide range of phenotypes caused by defects in the development and functioning of the central nervous system. Genetic factors play an important role in their aetiology. The view of the mode of genetic determination of neurodevelopmental disorders has changed several times during the last few decades. Today, a combined model prevails, according to which both common genetic variants with small effect and rare variants with strong effect participate in the development of these disorders, and both these mechanisms operate in parallel in each patient. Deciphering of polygenic networks of common variants is difficult, and the first associated loci have been reported only recently. Identification of variants with strong effect is easier, and they have been found in several hundred genes. Interestingly, these genes often support basic cellular processes, and their mutations appear to be tolerated in most systems, with the exception of the central nervous system. Progress in the identification of causal genes allows the definition of new syndromes involving neurodevelopmental disorders based on genetics. Today, however, this success is limited only to highly penetrant mutations in genes causing more frequent and more severe monogenic syndromes with lower phenotypic variability, and therefore many participating genes and the clinical entities linked to them remain unknown. Better understanding of the genetic mechanisms is already reflected in a more efficient genetic diagnostics, and will hopefully enable targeted therapy of some syndromes associated with neurodevelopmental disorders in the future. neurovývojová porucha, poruchy autistického spektra, vzácná varianta.
Key words: common variant, genetic heterogeneity, intellectual disability, neurodevelopmental disorder, autism spectrum disorders, rare variant
POJETÍ NEUROVÝVOJOVÝCH PORUCH (NDD)
Neurovývojové poruchy (neurodevelopmental disorders, NDD) jsou důsledkem abnormalit ve správném vývoji a fungování centrálního nervového systému. NDD jsou vymezeny v rámci DSM-5.1 Prvním projevem bývá opoždění psychomotorického vývoje dítěte, které vede k diagnóze mentální retardace (intellectual disability, ID). Mezi další fenotypové znaky (tučné písmo označuje první výskyt pojmu uvedeného v oddílu "Slovníček pojmů často používaných genetiky") patří opožděný vývoj řeči a řečové poruchy, poruchy autistického spektra (autism spectrum disorders, ASD), schizofrenie, porucha pozornosti s hyperaktivitou (ADHD), různé další abnormality chování, poruchy motoriky, a často i hypotonie a epilepsie (EPI). NDD jsou často asociovány s vrozenými vývojovými vadami a orgánovými dysfunkcemi různého charakteru a závažnosti, a s faciálním dysmorfismem. NDD postihují kolem 3 % dětí,2 a představují proto závažnou zátěž pro pacienty, jejich rodiny i celou společnost.
Většina fenotypových znaků klíčových pro NDD jsou znaky kvantitativní, pro které existuje v populaci spojité spektrum projevů. Kvantifikace znaků v rámci fenotypového spektra není vždy snadná a jednoznačná, a spektrum může mít i více dimenzí (např. endofenotypy u ASD, jako jsou problémy v sociální interakci, potíže s komunikací, tendence ke stereotypiím). Za patologii je považována určitá extrémní část spektra, jejíž hranice je obvykle definována dohodou. Biologicky však určitě existuje pro tyto znaky kontinuum.
Obecně není na přesné definici NDD úplná a trvalá shoda,3 viz i změny mezi DSM-4 a DSM-5. Někdy se v literatuře setkáváme s širším pojetím NDD zahrnujícím přesah do dalších neuropsychiatrických kategorií. Širší pojetí je podporováno sdílenou genetickou determinací některých znaků a jejich častou komorbiditou.4 Nejednotné je i používání samotného termínu NDD na hierarchické škále: někdy je míněn jeden fenotypový znak (endofenotyp), někdy samostatná choroba (vykazující několik nezávislých znaků), a někdy celý komplexní syndrom zahrnující znaky NDD. Ani následující text se určité terminologické promiskuitě nevyhne. Diskutované modely genetické etiologie však mohou obhájit širší pojetí NDD a také osvětlit rozpory, které u NDD dnes vznikají. Rozmlžení pojmů, kterého se zejména genetici dopouštějí, může být užitečné i pro uvědomění si obrovské variability NDD a pro pokus o jejich redefinici právě na základě genetiky.
Díky klesající ceně a narůstající dostupnosti se genetické analýzy již dnes provádějí u mnoha pacientů s těžšími nebo syndromovými formami NDD, a v budoucnu lze tuto praxi očekávat stále častěji. Genetickým vyšetřením mohou být pacienti přiřazeni ke konkrétním geneticky definovaným klinickým jednotkám. To umožňuje již dnes přesnější stanovení prognózy pacienta (porovnáním s dříve popsanými případy nesoucími podobný genetický defekt) a stanovení rizika rekurence onemocnění v rodině, v některých případech i cílenou léčbu. Nejen psychiatrické ambulance čeká doba, kdy budou pacienti stále častěji přicházet s genetickým výsledkem v ruce.
ROLE GENETIKY V ETIOLOGII NDD
Některé případy NDD mohou být podmíněny vnějšími vlivy (expozice chemickým látkám během těhotenství, perinatální poškození, úrazy, nádory, stres). Obecně však mají NDD výraznou genetickou komponentu, a především u těžších forem a forem s časným nástupem se předpokládá převážně genetická etiologie. Vlivy prostředí nelze odstínit a spolu s faktory epigenetickými se docela určitě podílejí na výsledném fenotypu každého pacienta. Nebudou však dále diskutovány a text se soustředí pouze na vliv genů a v nich se nacházejících variant, které tvoří různé alely těchto genů a tím formují genotyp sledovaného jedince.
Důležitost genotypu pro rozvoj NDD je podporována vysokými odhady heritability, získanými především u ASD ze studií dvojčat a dalších příbuzenských kombinací.5,6 Roli genetiky v rozvoji NDD podporuje také odedávna pozorovaná asociace NDD s mnohými genetickými defekty od chromozomálních (např. trizomie 21 [Downův syndrom]) až po genové (např. Rettův syndrom způsobovaný mutacemi genu MECP2 [gen pro metyl-CpG-vázající protein typu 2]).7 Ne všechny případy NDD se však dařilo přiřadit k některému ze známých způsobů monogenní dědičnosti, a především se donedávna u většiny pacientů s NDD nedařilo identifikovat jejich kauzální genetické defekty. To vedlo ke spekulacím o způsobech genetické determinace NDD.
GENETICKÉ MODELY A JEJICH MĚNÍCÍ SE PREFERENCE BĚHEM HISTORIE STUDIA NDD
Obecně existují pro choroby s výraznou genetickou komponentou dva základní modely genetické determinace. Jeden je postaven na populačně běžných, a druhý naopak na vzácných genetických variantách. Podle modelu mnoha běžných variant s malým účinkem vzniká u pacienta NDD kvůli nepříznivé kombinaci mnoha jednotlivě jen nepatrně škodlivých genetických variant. Ty jsou v populaci časté, ale nepostižení jedinci nesou pouze určitou část těchto variant, nikoliv kompletní nepříznivou kombinaci. Podle tohoto modelu by byly NDD polygenními či multifaktoriálními chorobami. Naopak model vzácných variant se silným účinkem předpokládá u každého postiženého pouze jednu nepříznivou variantu, která sama postačuje ke vzniku NDD; proto jsou tyto varianty vzácné a v nepostižené populaci se nevyskytují. NDD by tedy byly monogenními chorobami. Nepříznivé varianty by musely být při autozomálně dominantní dědičnosti vždy de novo nebo zděděné od postiženého rodiče, při autozomálně recesivní dědičnosti by je mohli mít i nepostižení přenašeči, a při X-vázané recesivní dědičnosti i nepostižené ženy - přenašečky.
Koncem 90. let převažoval u NDD (s výjimkou několika známých chromozomálních či monogenních forem) názor, že budou vysvětlitelné modelem mnoha běžných variant s malým účinkem. Pokusy identifikovat v genomu predisponující lokusy pomocí celogenomových vazebných a asociačních studií (genome-wide linkage/association studies, GWLS/GWAS) však byly opakovaně neúspěšné.
Po přelomu tisíciletí nastoupily technologie DNA čipů, od kterých se očekávalo, že podpoří GWAS díky své schopnosti paralelně genotypovat mnoho variant. Implementace metody však vedla k překvapivému objevu variant v počtu kopií (copy number variants, CNV) u pacientů s NDD. To obrátilo pozornost k modelu vzácných variant se silným účinkem. Objev CNV a potvrzení jejich asociace s NDD u mnoha pacientů (zejména tzv. "nové mikrodeleční syndromy")8 představovaly zásadní posun v chápání genetické determinace NDD.
Postupně se však ukázalo, že způsob dědičnosti některých CNV i jejich kauzalita pro NDD nebyly vždy přímočaré. Některé CNV byly zděděné od mírněji postiženého nebo zcela nepostiženého rodiče (neúplná penetrance) a klinický obraz postižených s danou CNV se často lišil - stejná varianta způsobovala různé NDD nebo různou tíži stejné NDD (variabilní expresivita). Tato pozorování nebyla monogenním modelem vysvětlitelná. Na rozdíl od zděděných variant nebyla také klasická lékařská genetika příliš zvyklá pracovat s de novo variantami.
Kolem roku 2010 došlo k širší implementaci metod sekvenování nové generace. Ty umožňovaly sekvenování celých exomů (exome sequencing, ES) nebo genomů (whole genome sequencing, WGS) pacientů s NDD. ES a WGS potvrdily a rozšířily poznatky získané na DNA čipech také na drobnější varianty na genové úrovni a postupně odhalily netušeně obrovskou genetickou heterogenitu NDD, která bude diskutována níže.
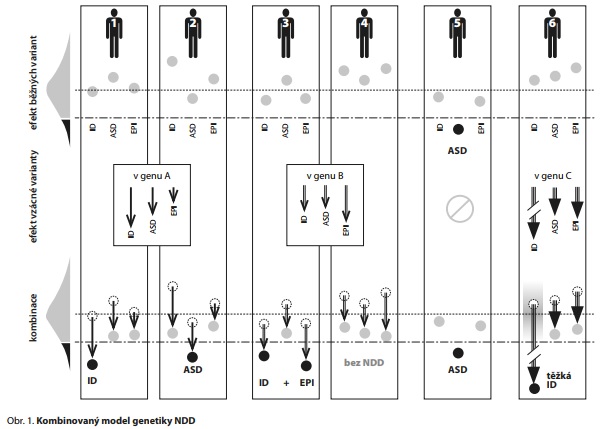
KOMBINOVANÝ MODEL GENETIKY NDD
Dnešní představa je taková, že oba modely působí u pacienta s NDD současně. Jakákoliv vzácná varianta se silným účinkem totiž existuje na konkrétním genovém pozadí sítě běžných variant, a oba typy efektu se navzájem ovlivňují.9-11 Kombinace obou modelů elegantně vysvětluje neúplnou penetranci i variabilní expresivitu.9 Častá de novo dědičnost vzácných variant vysvětluje častý sporadický výskyt NDD v rodinách a rozdíl ve frekvenci současného postižení mezi monozygotními a dizygotními dvojčaty (časté postižení obou monozygotních dvojčat, ale vzácné postižení obou dizygotních dvojčat - méně časté, než by vyplývalo ze způsobů monogenní dědičnosti).12 U různých NDD a u různých pacientů může být různý podíl příspěvku jednoho a druhého modelu, tedy vlivu běžných a vzácných variant.
Tento kombinovaný model ilustruje příklad šesti jedinců na obr. 1. Křivka vlevo nahoře představuje normální a patologickou část fenotypového spektra populační distribuce sledovaného znaku (šedě a černě) danou běžnými variantami, s populačním průměrem (tečkovaná horizontální čára) a diagnostickým prahem (čerchovaná horizontální čára). U každého pacienta jsou pro ilustraci sledovány tři znaky: úroveň kognitivních schopností, úroveň schopnosti sociální interakce/komunikace a aktivita mozku vyjádřená jako rezistence k EPI, jejichž pokles pod diagnostický práh vede k diagnóze ID, ASD či EPI. Úroveň těchto tří kvantitativních znaků je u každého jedince vyjádřena třemi kroužky, jejichž umístění výše či níže odráží vyšší či nižší úroveň znaku v normální nebo patologické části spektra (šedé nebo černé kroužky v horní třetině obrázku). U všech jedinců je vidět populační variabilita fenotypu, ale jen u jedince 5 postačuje vliv běžných variant k diagnóze ASD (tučně).
Vliv vzácné varianty, pokud se u jedince vyskytuje, je znázorněn dolů směřujícími šipkami, jejichž různá délka odráží různý negativní pleiotropní vliv varianty na tři sledované znaky (střední třetina obrázku). Negativní vliv na jednotlivé znaky je charakteristický pro každý gen. Jedinci 1 a 2 mají vzácnou variantu v genu A, která silně snižuje kognitivní schopnosti, mírněji poškozuje sociální interakci/komunikaci a ještě mírněji predisponuje k EPI. Mutace v genu B u jedinců 3 a 4 má jiný specifický vliv, podobně jako mutace v genu C u jedince 6, která ovšem vždy (na jakémkoliv pozadí běžných variant) těžce poškozuje kognici.
Kombinace vlivů běžných variant a vzácné varianty (dolní třetina obrázku) pak určuje výsledný stav sledovaných znaků u jedince. Stav zůstává v normální části spektra (šedé kroužky), nebo se posouvá do patologické části (černé kroužky) a vede k příslušné diagnóze (tučně).
U jedinců 1 a 2 podmiňují jejich běžné varianty různou úroveň sledovaných znaků, vždy v normální části spektra. Identická vzácná varianta s velkým účinkem v genu A, kterou oba nesou, vede u jedince 1 k diagnóze ID a u jedince 2 k diagnóze ASD (varianta v genu A tedy má variabilní expresivitu). Ostatní znaky jsou u obou také negativně ovlivněny, ale nedosahují diagnostického prahu.
U jedinců 3 a 4 nasedá vzácná varianta v genu B na kombinace běžných variant podmiňující podobně variabilní, nicméně opět normální úrovně sledovaných znaků, a u jedince 3 vede k diagnóze ID a EPI. U jedince 4 však výhodná (protektivní) kombinace běžných variant nedovolí projev varianty v genu B a jedinec 4 zůstává nepostižen (varianta v genu B tedy má neúplnou penetranci). To, že více genů (např. geny A a B) podmiňuje podobná, klinicky neodlišitelná postižení, označujeme jako lokusovou nebo jen genetickou heterogenitu. U některých genů záleží také na typu varianty či na jejím umístění v genu (alelová heterogenita).
Jedinec 5, jak je uvedeno výše, má diagnózu ASD podmíněnou pouze kombinací běžných variant a žádnou variantu se silným účinkem nenese. Konečně jedinec 6 by mohl být příkladem zrcadlové situace, kdy NDD způsobí samotná vzácná varianta. Pozadí běžných variant ovšem existuje vždy. Protože však mutace v genu C způsobuje tak závažný propad kognitivních schopností, že se projeví vždy, bez ohledu na výbavu běžnými variantami (při jakékoliv výchozí úrovni kognitivních schopností - šedá šmouha), můžeme zde běžné varianty ignorovat (úplná penetrance). Dnes dokážeme nalézat a asociovat s NDD většinou jen právě takové geny a varianty.
Pleiotropie zúčastněných genů způsobuje, že kromě tří výše sledovaných znaků může být ovlivněna i morfologie tváře, kterou nepříznivé běžné či vzácné varianty posouvají směrem k faciálnímu dysmorfismu, a další znaky typicky pozorované u NDD. Pleiotropie způsobuje i často pozorované přesahy NDD do dalších psychiatrických chorob.13 Naopak v jemnějším rozlišení můžeme podobně uvažovat o jednotlivých endofenotypech (např. u ASD mohou různé varianty různě ovlivňovat sociální chování, řeč, sklon ke stereotypiím).
Kombinovaný model ilustrovaný obr. 1 tedy na jedné straně, u výsledného efektu, podporuje existenci kontinua fenotypové variability pozorovaného v běžné populaci i v kohortách postižených NDD. Zdůrazňuje ovšem kontinuum i na straně druhé, u genotypů, které NDD podmiňují. Díky velké variabilitě, heterogenitě a multidimenzionalitě na obou stranách je každý jedinec v pravém slova smyslu zcela jedinečný, jak ve svém genotypu, tak v jím podmíněném fenotypu, včetně klinického obrazu NDD.
Ač může kombinovaný model působit komplikovaně, přesně odpovídá očekávání u systému pleiotropních genů, jejichž varianty mají různě závažný vliv na znaky spoluvytvářející fenotyp choroby, jsou-li tyto znaky současně kvantitativní a vesměs polygenně podmíněné. Klasická lékařská genetika to dosud měla jednodušší, protože se soustřeďovala na fenotypově jasně definované unikátní choroby, jako cystická fibróza nebo hemofilie A, které jsou podmíněné téměř výlučně penetrantními variantami s velkým účinkem v jednom svém řídícím genu.
DNEŠNÍ STAV POZNÁNÍ FENOTYPU A BĚŽNÝCH A VZÁCNÝCH VARIANT PODMIŇUJÍCÍCH NDD
Kategorizace fenotypu pacientů s NDD naráží na zmíněnou variabilitu a heterogenitu, a to i přes pokrok v diagnostických algoritmech a škálách a nástrojích pro posuzování jednotlivých znaků. Současně platí, že člověk je sociální primát, u kterého vnitrodruhová kompetice staví na schopnosti analyzovat duševní pochody a předvídat chování ostatních, a číst z morfologie a mimiky jejich tváří. Náš druh tedy má miliony let evoluce trvající historii učení intuitivního posuzování znaků, které jsou pro NDD nejdůležitější. Problémy s objektivizací faciálního dysmorfismu charakteristického pro některé genetické syndromy14 dokládají, že intuice zkušeného odborníka je klíčová a musí být respektována.
Snadná není pro jejich komplexitu ani analýza faktorů podmiňujících vznik NDD. V případě vnějšího prostředí může být obtížná např. detekce všech vlivů působících na jedince, kvantifikace každého vlivu a posouzení jeho rozložení v čase včetně časných vývojových stadií apod. Na podobné problémy naráží i epigenetika, např. na odlišnou metylaci různých pozic na chromozómech v různých buňkách jedince a na její změny během vývoje jedince. U genotypu by měla být analýza snazší pro jeho digitální povahu (v určité pozici chromozómu je buď nukleotid A, C, G, nebo T, zjednodušeně řečeno) a univerzalitu (všechny buňky jedince mají stejný genotyp, po celou dobu života, zjednodušeně řečeno). Problémem je ovšem např. to, že dnes (na konci roku 2020) je v lidském genomu popsáno téměř 700 milionů různých variant na genové úrovni, téměř 7 milionů CNV a podobných variant, a tím pádem i nevyčíslitelně obrovský počet jejich kombinací do konkrétních genotypů.
Zvláště obtížná je proto zejména identifikace polygenních sítí běžných variant podmiňujících NDD. Už jsme zmínili selhání GWLS a GWAS na přelomu tisíciletí. Teprve v roce 2019 bylo díky analýze větší kohorty pacientů a zlepšeným analytickým algoritmům odhaleno prvních pět lokusů asociovaných v polygenním modelu s ASD (na chromozómech 1, 7, 8 a 20 [2x]; někdy je signál přímo v zajímavých genech [KMT2E a MACROD2], jindy v jejich blízkosti).15 U NDD se tak stále potýkáme s problémem tzv. chybějící heritability ("missing heritability").16 Jde o stav, kdy se u choroby nedaří přes její prokázanou vysokou heritabilitu identifikovat pomocí GWAS jednotlivé přispívající varianty. To může být způsobeno účastí velkého množství běžných variant s velmi malým účinkem, nebo rolí vzácnějších variant, CNV a mezigenových interakcí, heterogenitou na úrovni polygenních sítí a dalšími komplikujícími faktory.
Mnohem jednodušší je, ovšem až díky ES a WGS, identifikace vzácných variant se silným účinkem. Ty byly u NDD v posledních letech nalezeny v mnoha různých genech (viz specializované databáze jako Decipher [https://decipher.sanger.ac.uk/ddd/ddgenes], SFARI [https://gene.sfari.org], DBD [https://dbd.geisingeradmi.org], Orphanet [http://www.orphadata.org/cgi-bin/index.php] a další). Analýzy podle modelu vzácných variant jsou dnes preferovány kvůli snazší identifikaci a interpretaci závažných variant ve srovnání s delikátnějšími variantami malého účinku, které mohou ležet např. v regulačních oblastech genů.15 I u modelu vzácných variant jsou ale dnes objevovány především vysoce penetrantní geny, které způsobují těžké a méně variabilní fenotypy.17 Naopak méně často mutované a méně penetrantní geny, varianty s jemnějšími účinky nebo varianty způsobující lehčí či variabilnější fenotypy zůstávají zatím neodhaleny.
Zdá se, že geny způsobující těžké monogenní NDD hrají roli i ve variabilitě kognitivních a behaviorálních rysů v normální populaci18 a že jeden gen může nést běžné varianty s malým účinkem i vzácné varianty se silným účinkem.15 Řeší se i otázka, zda se na různých NDD podílejí v různé míře běžné a vzácné varianty. Zdá se například, že těžké formy ID jsou převážně podmiňovány vzácnými variantami se silným účinkem, zatímco vysoce funkční formy ASD nebo lehké familiární formy ID, stejně tak jako obecně mírnější NDD v rodinách s více postiženými členy, mohou být determinovány převážně polygenně.15
REPERTOÁR GENŮ HRAJÍCÍCH ROLI V ROZVOJI NDD
Přes výše popsaný pokrok tedy dnes stále převládá studium protein-kódujících genů se vzácnými variantami se silným účinkem. Do jakých funkčních kategorií tyto geny spadají? Nepřekvapuje, že se často jedná o geny exprimované v mozku a podílející se na formování a funkci synapsí, neurální signalizaci a podobných procesech specifických pro nervový systém. Překvapivě velkou skupinu ale tvoří geny participující v těch nejzákladnějších buněčných procesech, jako jsou zajištění a regulace transkripce a translace genetické informace, modifikace chromatinu, metabolismus RNA, řízení stability proteinů, fungování mitochondrií a další podobné dráhy.
Tyto základní procesy probíhají ve všech buňkách a systémech organismu a je otázka, proč způsobují defekty těchto genů "pouze" NDD a nikoliv letalitu nebo těžká multisystémová postižení. Odpověď může být v celkové robustnosti vývoje lidského jedince, díky které je mnoho genových defektů tolerováno a neprojeví se negativně na fenotypu. Tato tolerance ale nemusí fungovat právě u znaků, které jsou evolučně nejmladší, nejpokročilejší, a tedy zřejmě nejfragilnější, jako jsou lidské kognitivní schopnosti a komplexní vzorce sociálního chování, pro jejichž rozvoj je nutný zcela nenarušený vývoj mozku. Defekty ve zmíněných genech se tedy mohou projevit jen fenotypovými příznaky charakteristickými pro NDD.19 Rozdíly v robustnosti vývoje mezi pohlavími zřejmě podmiňují pozorovaný vyšší výskyt většiny NDD u chlapců, tzv. "female-protective effect". Vývojová povaha mnoha těchto genů je můstkem k pozorovaným odchylkám v morfologii tváře. A ukazuje se také (a může to být i vodítkem při odhalování nových kauzálních genů), že somatické mutace či deregulace mnohých těchto genů způsobují nádorová onemocnění.
Zajímavá je i otázka celkového počtu protein-kódujících genů podmiňujících monogenní NDD. Odhady jsou předmětem diskuse a jsou značně variabilní, ale naznačují existenci několika tisíc, možná až pěti tisíc takových genů.20-22 Genetická heterogenita monogenních forem NDD je tedy obrovská. Takový počet tvoří velmi podstatnou část z celkového počtu genů v lidském genomu (zhruba 20 000). Přestože si dovedeme představit, že pokročilé funkce lidského mozku vyžadují značnou genovou podporu, a přestože je známo, že v mozku je ve srovnání s jinými orgány exprimován největší podsoubor genů, je tento počet překvapivě vysoký. A mohlo by se zdát, že nemusí zbývat dost genů na ostatní orgánově-specifické procesy a na jiné choroby než NDD. Tento paradox může být nejspíše opět vysvětlen pleiotropií genových účinků. Např. gen BCL11A (kódující část chromatin remodelačního komplexu BAF) se podílí na rozvoji monogenní NDD - Diasové-Loganova syndromu, současně ovlivňuje perzistenci fetálního hemoglobinu a hraje roli v několika nádorových onemocněních.23
Znalost aspoň části repertoáru genů asociovaných s NDD také dovoluje zkoumat, do jaké míry mohou být jednotlivé geny specifické pro jednu nebo druhou NDD, avšak ani zde zatím nejsou názory jednotné.24,25
DŮSLEDKY ZNALOSTÍ O GENETICKÉ DETERMINACI NDD PRO JEJICH DEFINICI A DIAGNOSTIKU
Kvůli limitacím v našem poznání jsme tedy dnes schopni laboratorně diagnostikovat a tím i vzájemně odlišit pouze monogenní NDD podmíněné vzácnými variantami se silným účinkem a vysokou penetrancí, spojené s těžšími fenotypy. Tak byly v posledních letech popsány stovky nových klinických jednotek definovaných svým kauzálním genem, převažujícím typem variant a fenotypem zahrnujícím znaky NDD. Díky obrovské genetické heterogenitě, podmíněné tisíci kauzálních genů, jsou jednotlivé monogenní NDD velmi vzácné. Jde tedy o obrovskou skupinu vzácných onemocnění s podobnými projevy.
To ovšem způsobuje, že pro mnoho nových klinických jednotek bylo popsáno jen několik pacientů po celém světě, nejvýše desítky či nízké stovky. A ačkoliv byly mnohé z těchto syndromů deklarovány jako klinicky rozpoznatelné (obvykle podle faciálního dysmorfismu a dalších znaků), nedostatek opravdu specifických symptomů a fenotypová variabilita pacientů kombinované se vzácností syndromu a nemožností získat diagnostické zkušenosti na větším souboru obvykle neumožňují diagnostiku jednotlivých geneticky definovaných NDD na základě fenotypu. Pro dosažení diagnózy proto musí být použit přístup "genotype-first", tj. vyjít z identifikace konkrétního genetického defektu.26 Pak musí následovat podrobné fenotypování pacienta a posouzení jeho podobnosti s dříve popsanými případy s variantami v daném genu. Problém nedostatečné specificity symptomů v kombinaci s jejich variabilitou mezi pacienty se samozřejmě týká i jednotlivých endofenotypů, takže ač to je teoreticky možné (a bylo to uvedeno v diskusi kombinovaného modelu), v praxi může být obtížné posoudit, zda a jak se liší klinický obraz např. ASD u jednotlivých genů.
Podobně je třeba postupovat při podezření na novou, dosud nepopsanou NDD. Nejdříve je třeba zjistit, zda někdo na světě nezachytil pacienty s variantou v témže genu, a porovnat jejich fenotypy. Pokud je shoda v typu variant a ve fenotypu, pokud se podobné varianty nenacházejí u nepostižených, pokud je dotčený gen zajímavý svou funkcí nebo tkáňově-specifickou expresí, a obvykle i po průkazu negativního vlivu variant na funkci genu a jeho proteinového produktu (tzv. funkční studie), je možné popsat novou klinickou jednotku.
Nové technologie celogenomové analýzy a výše nastíněný postup umožnily identifikaci genetických variant u mnoha pacientů, u kterých se to dříve klasickými metodami nedařilo, a u kterých až tyto postupy ukončily často mnohaletou "diagnostickou odyseu". Vysoká incidence NDD obecně a jejich velká genetická heterogenita často způsobují, že i dva podobně postižení sourozenci mohou nést varianty v různých genech, a patřit tedy do různých klinických jednotek. Identifikace kauzální varianty je prakticky cenná pro lepší prognózu, informované rozhodování o reprodukci a často i jistou úlevu v rodině z ukončení nejistoty. Přes veškerý pokrok však zůstává neobjasněna zhruba polovina pacientů, u kterých se předpokládá genetická etiologie jejich NDD.27
ZÁVĚR
Přestože může výše uvedené vyvolat určitou skepsi, je třeba kvitovat obrovský pokrok, ke kterému během několika posledních let došlo. Genetika NDD dnes zažívá velice vzrušující dobu, ve které je často potkávaná nejistota vykoupena možností přispět k odhalení nových genů, syndromů a genetických mechanismů. Pokrok ve výzkumu se ovšem rychle přetavuje do lepších možností genetické diagnostiky.
I díky práci několika velkých výzkumných konsorcií (např. Deciphering Developmental Disorders [https://www.ddduk.org] nebo Autism Sequencing Consortium [https://genome.emory.edu/ASC]) lze v blízké budoucnosti očekávat pokrok v porozumění sítím běžných variant podílejících se na NDD, protože dnešní GWAS začínají využívat všechny varianty odhalené ES nebo zejména WGS, nikoliv pouze vybrané časté varianty testované na DNA čipech. Toto je kombinováno s analýzou zvětšujících se kohort a s využitím lepších analytických algoritmů.
Lze také očekávat kompletaci katalogu genů zodpovědných za monogenní NDD, postupně včetně vzácnějších a méně penetrantních forem. I zde jsou k dispozici stále lepší nástroje, plnící se databáze patogenní a normální variability (dnes již u statisíců normálních jedinců),28 lepšící se informace o jednotlivých genech (odolnost vůči variabilitě, exprese ve fetálním mozku, funkce, interakční partneři), dostupnější funkční studie apod. Sice pomalu, ale přece jen rostoucí počty pacientů s variantami v jednotlivých genech také umožňují přesnější definici fenotypového obrazu jednotlivých monogenních NDD.
Zlepší se jistě i naše znalosti o dalších genetických mechanismech potenciálně hrajících roli u NDD, jako jsou varianty v regulačních oblastech genů a v genech pro funkční RNA (nekódující geny), či mechanismech zahrnujících mozaicismus nebo mobilní DNA apod.
Kromě diagnostiky je samozřejmě důležitá i léčba NDD. Obecně existuje prakticky u všech genetických chorob obrovská prodleva mezi diagnostikou a terapií, ale i zde lze opatrně očekávat pokrok (opatrně i proto, že vývojová povaha mnoha NDD a priori nedává nejlepší vyhlídky na kauzální léčbu započatou po projevení se choroby, a také půjde kvůli vysoké genetické heterogenitě NDD o léčbu vzácných onemocnění se všemi jejími problémy). Identifikace konkrétních genů a genetických mechanismů je ovšem nutnou podmínkou a prvním krokem k případné budoucí kauzální léčbě.29,30
Slovníček pojmů často používaných genetiky
alela - forma genu daná většinou variantami v jeho sekvenci celogenomové vazebné/asociační studie (genome-wide linkage/association studies, GWLS/GWAS) - postupy zjišťující, zda jsou určité alely přednostně předávány postiženým dětem v rodině, resp. zda se určitá alela vyskytuje častěji v kohortě nepříbuzných postižených ve srovnání s kohortou nepostižených
de novo - genetická varianta přítomná u dítěte, ale ne u žádného z jeho rodičů
DNA čip - většinou pevný povrch s navázaným velkým množstvím různých sekvencí DNA, který umožňuje např. zjištění genotypu nebo počtu kopií v mnoha místech genomu najednou epigenetika - ovlivnění exprese genů prostřednictvím modifikací chromatinu nebo regulačních nekódujících RNA exom - soubor exonů (částí genů, které jsou součástí mRNA) všech protein-kódujících genů
fenotyp - soubor všech vlastností jedince, zjistitelných přímým pozorováním nebo specializovanými vyšetřovacími metodami
gen - úsek molekuly DNA, který kóduje protein (protein-kódující geny) nebo funkční RNA (nekódující geny) genetická heterogenita - stav, kdy je stejný znak/soubor znaků (včetně choroby) podmíněn alternativně variantami v různých genech (lokusová heterogenita), popř. různými variantami v jednom genu (alelová heterogenita)
genom - soubor veškeré DNA organismu včetně všech genů genotyp - konkrétní alelová výbava jedince, buď v jednom genu, nebo ve všech genech
heritabilita - podíl genetických faktorů na variabilitě znaku, zjednodušeně vyjádřený zlomkem (vliv genetických faktorů) / (vliv genetických faktorů + vliv prostředí)
lokus - místo na chromozómu, obsazené genem, regulační sekvencí, variantou apod.
mobilní DNA - úseky molekuly DNA, které se mohou v genomu samovolně přesouvat/zmnožovat
mozaicismus - jedinec je tvořen více buněčnými liniemi s různým genotypem, které pocházejí z jedné zygoty mutace - genetická varianta s výskytem v populaci nižším než 1 %, často s negativním vlivem na fenotyp; genetici nedoporučují termín používat, přesto se málokterá učebnice nebo odborný text bez tohoto pojmu obejdou
neúplná penetrance - stav, kdy se genetické postižení nemusí u jedince s variantou projevitpleiotropie - schopnost (většiny lidských) genů ovlivňovat několik znaků, vykonávat více funkcí
sekvenování nové generace (next generation sequencing, také masivně paralelní sekvenování) - metody umožňující zjištění nukleotidové sekvence mnoha různých fragmentů DNA najednou
variabilní expresivita - stav, kdy se spektrum a tíže příznaků mohou lišit mezi jedinci se stejnou variantou varianta (genetická) - odchylka od referenční sekvence lidského genomu
varianty na genové úrovni - varianty postihující méně než 50 nukleotidů, buď jednonukleotidové varianty (single nucleotide variants, SNV, nebo single nucleotide polymorphisms, SNP), nebo krátké delece/inserce (indel)
varianty v počtu kopií (copy number variants, CNV) - delece nebo duplikace/amplifikace zahrnující více než 50 nukleotidů; CNV a podobně velké varianty, nezpůsobující zisk/ztrátu genetického materiálu, se souhrnně nazývají strukturní varianty
LITERATURA
- 1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed.). Arlington: APA 2013.
- 2. Lopez-Rivera JA, Perez-Palma E, Symonds J et al. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain 2020; 143 (4): 1099-1105.
- 3. Ismail FY, Shapiro BK. What are neurodevelopmental disorders? Curr Opin Neurol 2019; 32 (4): 611-616.
- 4. Coe BP, Girirajan S, Eichler EE. The genetic variability and commonality of neurodevelopmental disease. Am J Med Genet C Semin Med Genet 2012; 160C (2): 118-129.
- 5. Constantino JN, Todorov A, Hilton C et al. Autism recurrence in half siblings: strong support for genetic mechanisms of transmission in ASD. Mol Psychiatry 2013; 18 (2): 137-138.
- 6. Rosenberg RE, Law JK, Yenokyan G et al. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med 2009; 163 (10): 907-914.
- 7. Havlovicova M, Sedlacek Z. Genetika autismu. In: Hrdlicka M, Komarek V (eds). Dětský autismus. Praha: Portal 2014: 129-170.
- 8. Weise A, Mrasek K, Klein Eet al. Microdeletion and microduplication syndromes. J Histochem Cytochem 2012; 60 (5): 346-358.
- 9. Gonzalez-Mantilla AJ, Moreno-De-Luca A, Ledbetter DH, Martin CL. A Cross-Disorder Method to Identify Novel Candidate Genes for Developmental Brain Disorders. JAMA Psychiatry 2016; 73 (3): 275-283.
- 10. Iakoucheva LM, Muotri AR, Sebat J Getting to the Cores of Autism. Cell 2019; 178 (6): 1287-1298.
- 11. Kurki MI, Saarentaus E, Pietilainen Oet al. Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland. Nat Commun 2019; 10 (1): 410.
- 12. Beaudet AL Autism: highly heritable but not inherited. Nat Med 2007; 13 (5) : 534-536.
- 13. Lee PH, Feng YA, Smoller JW. Pleiotropy and Cross-Disorder Genetics Among Psychiatric Disorders. Biol Psychiatry 2021; 89 (1): 20-31.
- 14. Hallgrimsson B, Aponte JD, Katz DC et al. Automated syndrome diagnosis by three-dimensional facial imaging. Genet Med 2020; 22 (10): 16821693.
- 15. Grove J, Ripke S, Als TD et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 2019; 51 (3): 431-444.
- 16. Manolio TA, Collins FS, Cox NJ et al. Finding the missing heritability of complex diseases. Nature 2009; 461 (7265): 747-753.
- 17. Kaplanis J, Samocha KE, Wiel L et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 2020; 586 (7831): 757-762.
- 18. Price KM, Wigg KG, Feng Yet al. Genome-wide association study of word reading: Overlap with risk genes for neurodevelopmental disorders. Genes Brain Behav 2020; 19 (6): e12648.
- 19. Suliman R, Ben-David E, Shifman S. Chromatin regulators, phenotypic robustness, and autism risk. Front Genet 2014; 5: 81.
- 20. Coe BP, Stessman HAF, Sulovari A et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet 2019; 51 (1): 106-116.
- 21. Jamra R. Genetics of autosomal recessive intellectual disability. Med Genet 2018; 30 (3): 323-327.
- 22. Neri G, Schwartz CE, Lubs HA, Stevenson RE. X-linked intellectual disability update 2017. Am J Med Genet A 2018; 176 (6): 1375-1388.
- 23. Dias C, Estruch SB, Graham SA et al. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am J Hum Genet 2016; 99 (2): 253-274.
- 24. Myers SM, Challman TD, Bernier R et al. Insufficient Evidence for "Autism-Specific" Genes. Am J Hum Genet 2020; 106 (5): 587-595.
- 25. Schaaf CP, Betancur C, Yuen RKC et al. A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat Rev Genet 2020; 21 (6): 367-376.
- 26. Stessman HA, Bernier R, Eichler EE. A genotype-first approach to defining the subtypes of a complex disease. Cell 2014; 156 (5): 872877.
- 27. Lee H, Nelson SF. The frontiers of sequencing in undiagnosed neurodevelopmental diseases. Curr Opin Genet Dev 2020; 65: 76-83.
- 28. Karczewski KJ, Francioli LC, Tiao G et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581 (7809): 434443.
- 29. Malinowski J, Miller DT, Demmer L et al. Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet Med 2020; 22 (6): 9861004.
- 30. Levy G, Barak B. Postnatal therapeutic approaches in genetic neurodevelopmental disorders. Neural Regen Res 2021; 16 (3): 414-422.