Česká a slovenská psychiatrie

Časopis
Psychiatrické společnosti ČLS JEP
a Psychiatrickej spoločnosti SLS
souborný článek / review article
- AKTUÁLNÍ ČÍSLO
- ARCHIV
- VYHLEDÁVÁNÍ
- PERIODIKUM
- REDAKČNÍ RADA
- PŘEDPLATNÉ
- INZERCE
- AKTUALIZOVANÉ
POKYNY
PRO AUTORY
- ISSN 1212-0383




- © Česká a Slovenská psychiatrie 2025
- © Galén 2025
PSYCHIATRICKÁ MANIFESTACE DĚDIČNÝCH METABOLICKÝCH PORUCH
PSYCHIATRIE MANIFESTATION OF INBORN ERRORS OF METABOLISM
Jan Kulhánek1, Jakub Albrecht2, Tomáš Honzík1, Martin Magner1
1 Klinika dětského a dorostového lékařství 1. LF UK a VFN, Praha
2 Psychiatrická klinika 1. LF UK a VFN, Praha
2 Psychiatrická klinika 1. LF UK a VFN, Praha
Práce byla podpořena granty PRVOUK-P24/LF1/3, PRVOUK-P26/LF1/4, UNCE 204011 a RVO-VFN 64165/2012.
SOUHRN
Kulhánek J, Albrecht J, Honzík T, Magner M. Psychiatrická manifestace dědičných metabolických poruch
Dědičné metabolické poruchy (DMP) tvoří heterogenní skupinu více než 1000 nemocí s extrémní klinickou variabilitou a tíží manifestace. Tato onemocnění se často manifestují již v dětství, avšak zejména poslední dobou byla popsána řada lehčích a pozdně se manifestujících forem nezřídka s různou psychiatrickou manifestací (PM). Jedná se o onemocnění vzácná, klinická zkušenost s konkrétní poruchou bývá malá. Cílem práce je nastínit problematiku PM u pacientů s DMP, přiblížit principy patofyziologie DMP a na příkladu kazuistik vybraných onemocnění zvýšit povědomí o této skupině nemocí. Varovným příznakem PM u DMP může být familiární výskyt a časný rozvoj onemocnění, nápadná fluktuace klinických příznaků, kognitivní deteriorace, zrakové halucinace, přítomnost katatonie, rezistence na běžnou léčbu, vyšší četnost nežádoucích účinků terapie, či dokonce zhoršení PM po jejím zahájení. Klasickými příklady takovýchto onemocnění jsou poruchy cyklu močoviny, poruchy metabolismu sirných aminokyselin, akutní porfyrie, Niemannova-Pickova choroba typ C, Tayova-Sachsova nemoc, adultní forma metachromatické leukodystrofie, X-vázaná adrenoleukodystrofie, Wilsonova choroba a mitochondriální onemocnění. Diagnostika je složitá a měla by být provedena ve spolupráci se specializovaným centrem. Její význam je zdůrazněn dostupností účinné terapie pro řadu z těchto onemocnění.
Klíčová slova: dědičné metabolické onemocnění, poruchy cyklu močoviny, Niemannova-Pickova choroba typ C
SUMMARY
Kulhánek J, Albrecht J, Honzík T, Magner M. Psychiatrie manifestation of inborn errors of metabolism
Inborn errors of metabolism (IEM) comprise a heterogeneous group of more than 1000 diseases with extreme clinical variability and severity of manifestation. These disorders often present themselves mainly in childhood. However, a number of less severe and later-onset forms often with a varied psychiatric manifestation (PM) has been described lately. Since the diseases are rare, clinical experience with particular disorder is usually diminutive. The aim of this work is to shed light upon the issue of PM in patients with IEM, to outline the principles of the pathophysiology of IEM and to raise awareness of this group of disorders upon examples of selected diseases case reports. Cautionary signs of PM of IEM can be familial occurence and early onset of the disease, conspicious fluctuation of clinical signs, cognitive deterioration, visual hallucinations, presence of catatonia, resistance to common treatment, higher frequency of adverse events of the therapy or even worsening of PM after its commencement. Typical examples of such disorders are urea cycle defects, disorders of sulfur amino acids metabolism, acute porphyrias, Niemann-Pick disease type C, Tay-Sachs disease, adult form of metachromatic leukodystrophy, X-linked adrenoleukodystrophy, Wilson disease and mitochondrial diseases. The diagnostics is intricate and ought to be conducted in cooperation with a specialised centre. Its importance is emphasized by the availability of effective therapy for a number of these disorders.
Key words: inborn errors of metabolism, urea cycle disorders, Niemann-Pick disease type C
ÚVOD
Dědičné metabolické poruchy (DMP) tvoří heterogenní skupinu více než 1000 nemocí. Patofyziologickým podkladem DMP je ve většině případů porucha biochemické reakce daná špatně fungujícím enzymem na základě mutace v nukleární nebo mitochondriální DNA. I když se jedná o onemocnění vzácná, jejich souhrnný výskyt se odhaduje minimálně na 1 : 500.1 Extrémně široká je klinická variabilita onemocnění od závažné manifestace v časném věku se závažnou prognózou až po oligosymptomatické formy diagnostikované v pozdním věku (obr. 1). Právě v posledně jmenované skupině narůstá i počet kazuistik pacientů s DMP, u kterých se onemocnění manifestovalo psychiatrickými poruchami. Řada DMP je přitom poměrně dobře léčitelných a nestanovení diagnózy může mít stran prognózy pro pacienta zásadní význam.
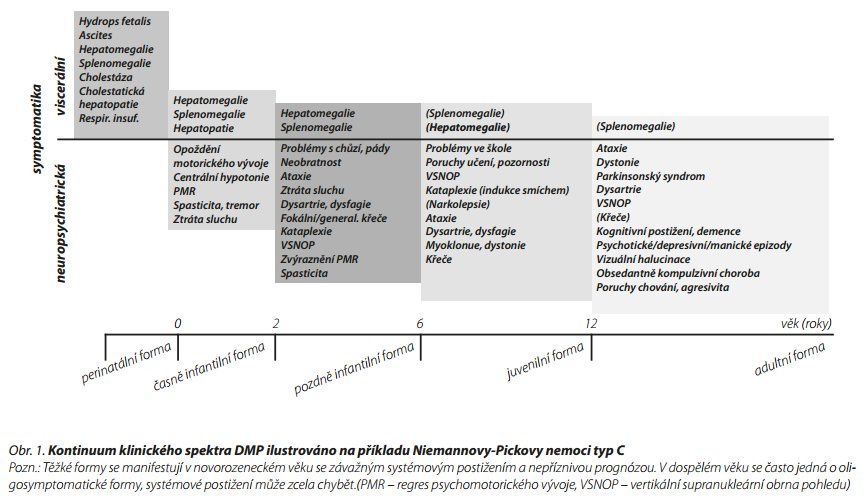
Na patofyziologii psychiatrické manifestace (PM) DMP se podílí zejména koncept akutního toxického působení malých molekul (jako například amoniaku), pomalá akumulace velkých komplexních molekul u lysosomálních onemocnění či nedostatek glukózy a ATP u DMP energetického metabolismu.1 Velmi důležité z pohledu psychiatrických symptomů DMP je však i narušení myelinizace kortiko-subkortikálních drah a kortiko-kortikálních drah (zejména dráhy fronto-temporální) (například u adultní metachromatické leukodystrofie, X-vázané adrenoleukodystrofie a Niemannovy-Pickovy nemoci typ C). Postižení fronto-temporální dráhy je přitom dáváno do souvislosti s rozvojem schizofrenie.2 Ovlivnění množství dendritů a míry dendritické interkomunikace mezi neurony může vysvětlit některé psychiatrické symptomy u pacientů s Tayovou-Sachsovou nemocí a Niemannovou-Pickovou nemocí typ C. Na úrovni neurotransmiterů (porucha metabolismu biogenních aminů, nadprodukce receptorů pro biogenní aminy, nadbytek glutaminergní transmise, GABA deficience) působí například akutní porfyrie a fenylketonurie.
V následujícím přehledu jsme si proto dali za úkol čtenáře stručně obeznámit s problematikou nejčastěji uváděných DMP s psychiatrickou manifestací z pohledu metabolického specialisty. Teoretický úvod je pro ilustraci doplněn krátkými kazuistikami z odborné literatury nebo našich zkušeností.
Pro přehlednost jsme DMP metodicky rozdělili do tří skupin:
1. DMP malých molekul, často doprovázené akutním metabolickým rozvratem,
2. DMP velkých neboli komplexních molekul, s chronickým progresivním průběhem,
3. DMP energetického metabolismu, tedy mitochondriální onemocnění.
Tyto skupiny se liší stran patofyziologie i průběhu psychiatrické manifestace. V následujícím textu jsou jednotlivé skupiny probrány podrobněji.
NEMOCI MALÝCH MOLEKUL
Jedná se o poruchu intermediárního metabolismu malých molekul, tj. s molekulovou hmotností do 1500 daltonů. Nezpracovaný substrát se hromadí a stává se pro organismus toxický. Rozvíjí se metabolický rozvrat s klinickými a laboratorními projevy závislými na konkrétní toxické látce, často doprovázený poruchou acidobazické rovnováhy a homeostázy glykémie. Principem akutní i chronické léčby těchto onemocnění je předcházení metabolickému rozvratu omezením zvýšené nabídky nedostatečně zpracovatelných chemických látek původu exogenního (např. dietní omezení živin, které se u dané DMP špatně metabolizují, např. bílkovin u poruch cyklu močoviny), ale i endogenního, a to dostatečným a pravidelným energetickým příjmem jako prevencí katabolického stavu. Jako klasické příklady onemocnění prezentujících se psychiatrickou manifestací lze z této skupiny uvést poruchy cyklu močoviny (amoniak), akutní porfyrie (kyselina δ-aminolevulová) a dědičnou formu homocystinurie (homo cystein).
Poruchy cyklu močoviny
Vedlejším produktem metabolismu dusíkatých látek, především bílkovin, je amoniak, jehož hladina v krvi je za normálních okolností < 60 µmol/1 (u novorozenců a kojenců < 80 µmol/1). Homeostázu amoniaku zajišťuje cyklus močoviny, jehož enzymy metabolizují amoniak na močovinu. Zvýšená koncentrace amoniaku působí neurotoxicky, hepatotoxicky a nefrotoxicky V souvislosti s psychiatrickou manifestací u dospělých pacientů se zmiňuje zejména X-vázaný deficit ornitintranskarbamylázy (OTC), který se však často manifestuje i u dívek a žen.3 Ostatní poruchy jsou děděny autosomálně recesivně. Onemocnění se může manifestovat poruchou vědomí (hyperamonemické koma) až s 50% letalitou. Přidružují se respirační alkalóza, křeče, jaterní selhání, později hypotonie, neprospívání, zvracení, růstová retardace a opoždění vývoje. Náhlému propuknutí psychiatrických příznaků může předcházet operační výkon, porod či trauma, které vyvolají katabolismus proteinů a následnou hyperamonemickou encefalopatii. Toto se týká zejména do té doby asymptomatických žen přenašeček deficitu OTC.
U 21leté ženy se osm dní po porodu objevily bolesti hlavy, zmatenost a přestala komunikovat. Po přijetí na psychiatrickou kliniku byla určena diagnóza poporodní deprese. Do 24 hodin po přijetí (11 dní po porodu) se rozvinulo koma s edémem papil, tonicko-klonické křeče a dekortikační postura. Byla zjištěna významná hyperamonémie (411 µmol/l). Žena byla úspěšně léčena hemodialýzou a i. v. natrium benzoátem a arginin hydrochloridem. Byl diagnostikován deficit OTC.4
Porfyrie
Hereditární porfyrie jsou skupinou osmi metabolických poruch biosyntézy hemu. Psychiatrické příznaky se vyskytují u akutních forem, které se dále vyznačují kolikovitými bolestmi břicha a neurologickými příznaky. Porfyrické ataky mohou být vyvolány infektem, léky, alkoholem, zátěží, menstruací a lačněním. Klinické příznaky mohou začínat prodromální fází zahrnující změny chování, jako úzkost, neklid a nespavost. Prakticky konstantní je těžká bolest břicha, častá je nauzea, zvracení, zácpa a doprovázející známky aktivace sympatiku - tachykardie, pocení, hypertenze. Ataka může být komplikována dehydratací a hyponatrémií, která může vést až ke křečím. Někdy se může vyskytnout tmavě zbarvená moč. Jednotlivé ataky bývají většinou v trvání dnů až několika týdnů.5 Až polovina pacientů má psychiatrické příznaky. Může se jednat o psychotické epizody, ale také depresi, úzkost a delirium, které mohou být prvními příznaky. Intermitentní psychotické epizody mohou vést až k diagnóze schizofrenie. Pacienti mohou dokonce být pro břišní bolest vyvolanou akutní zátěží diagnostikováni se somatoformní poruchou nebo histriónskou poruchou osobnosti.6
15letá dívka s rok diagnostikovanou schizofrenií byla hospitalizována pro dva dny trvající stav spojený s bizarním chováním. Její slovní projev byl rozvolněný až tangenciální, myšlení bylo inkoherentní, obsahově bludně paranoidní, své spolužáky označovala za mimozemšťany. Před zmíněnou epizodou fungovala dobře jak ve škole, tak v sociálních interakcích při mnoha aktivitách. Od první hospitalizace prodělala čtyři méně závažné ataky, kdy se "chovala a mluvila legračně", nicméně její chování nebylo natolik nápadné, aby byla odvezena do nemocnice. Epizodám předcházel mírný infekt horních cest dýchacích s teplotou a jinak tomu nebylo ani v tomto případě. Podezření na ataky akutní porfyrie bylo podpořeno jantarovou barvou moči. Diagnóza akutní intermitentní porfyrie byla stanovena potvrzením zvýšené exkrece kyseliny δ-aminolevulové a porfobilinogenu v moči. Terapie haloperidolem a quetiapinem byla ukončena, změněna na chlorpromazin. S rodinou byla probrána režimová opatření, dívka byla po následujících 12 měsíců v pořádku.7
Homocystinurie
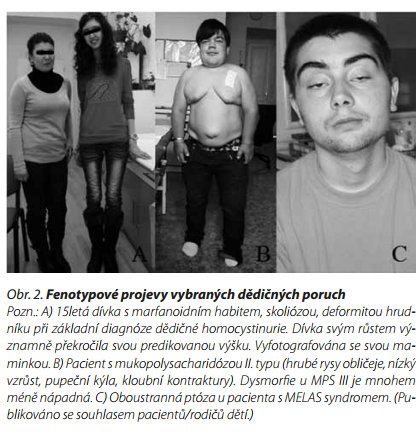
Homocystinurie z deficitu cystathionin-β-syntázy je nejčastějším typem poruchy metabolismu methioninu. Klinicky se pacienti projeví postižením CNS (opoždění vývoje, regres, epilepsie; spíše u časné manifestace), pojiva (marfanoidní habitus sub/luxace oční čočky (obr. 2) a vaskulárními příznaky (trombózy, cévní mozkové příhody; manifestace spíše u adultních případů). Léčba směřující k snížení hladiny homocysteinu zahrnuje nízkobílkovinovou dietu se suplementací esenciálních aminokyselin a v případě responzivních forem podávání vysokých dávek pyridoxinu.8 V souboru 63 pacientů s homocystinurií se psychiatrické příznaky vyskytly v 51 %.9 Nejčastěji se jednalo o epizodické deprese (10 %), chronické poruchy chování (17 %), chronickou obsesivně-kompulzivní poruchu (5%) a poruchy osobnosti (19 %). Průměrné IQ bylo 80 ? 27. Agresivní chování a poruchy chování se vyskytovaly zvláště u pacientů s opožděním vývoje a pacientů neodpovídajících na léčbu pyridoxinem. Byly však publikovány i izolované případy pacientů s psychózou.9 Na diagnózu lze vyslovit podezření u pacientů s vysokou koncentrací homocysteinu a methioninu v krvi.
Psychiatrické projevy charakteru akutního stavu dezorientace, letargie a úzkosti byly popsány i u třetiny pacientů s remethylační formou homocystinurie na podkladě deficitu enzymu methylentetrahydrofolát reduktázy (MTHFR).10
CHRONICKÝ PROGRESIVNÍTYP DMP
Ilustrativním příkladem je skupina více než 70 známých lysosomálních onemocnění, při kterých dochází nejčastěji při nedostatečné aktivitě některého z lysosomálních enzymů k střádání nedegradovaných komplexních molekul. To pak spouští řadu sekundárních patofyziologických pochodů, např. změnu buněčné signalizace, aberantní genovou expresi, zvýšenou apoptózu. Na rozdíl od první skupiny akutně intoxikačních onemocnění se jedná o onemocnění chronicky progresivní, při kterých nedochází k metabolickým krizím neboli rozvratům. Mezi onemocnění s psychiatrickou manifestací lze z této skupiny uvést mukopolysacharidózu typ II (Hunterův syndrom), typ III (Sanfilippo syndrom). Dalším příkladem je Niemannova-Pickova nemoc typ C, gangliosidóza GM2 (Tayova- Sachs ova nemoc) nebo metachromatická leukodystrofie. Vzhledem k chronickému průběhu onemocnění v této části uvedeme i X-vázanou adrenoleukodystrofii a Wilsonovou nemoc.
Mukopolysacharidózy
Mukopolysacharidózy jsou skupinou lysosomálních střádavých onemocnění, která se vyznačují hromaděním glykosaminoglykanů (dříve mukopolysacharidů). Výsledkem je multisystémové progresivní postižení, pro které jsou typické zejména hrubé rysy obličeje, hepatosplenomegalie, nízký vzrůst, změny skeletu charakteru dysostosis multiplex, postižení srdečních chlopní a často postižení CNS, zejména ve smyslu postižení psychomotorického vývoje. Psychiatrickou manifestací se vyznačuje zejména Hunterův syndrom (mukopolysacharidóza typ II) a Sanfilippo syndrom (mukopolysacharidóza typ III). Diagnóza na biochemické úrovni je založena na stanovení mukopolysacharidů v moči (nejlépe z 12hodinového sběru), podezření je však nutné potvrdit enzymatickým vyšetřením v leukocytech a molekulárně-genetickým vyšetřením.
Mukopolysacharidóza typ II, Hunterův syndrom
Jedná se o X-vázané onemocnění, takže postiženi jsou prakticky pouze chlapci (obr. 2). Postižení CNS je přítomno cca ve dvou třetinách případů a odpovídá těžkému fenotypu onemocnění. Pacienti s mírnou formou mohou mít relativně normální mentální vývoj, řeč není opožděna nebo pouze mírně. Sami jsme léčili dospělého pacienta s ukončeným postgraduálním vzděláním.11 U chlapců s těžkou formou dochází po postupném opožďování zejména v batolecím věku k vývojovému plateau a regresu nejčastěji mezi 48. a 60. měsícem života. Mírnější forma onemocnění se neváže s poruchou chování, zatímco u závažného fenotypu jsou tyto poruchy prakticky konstantní. Nejčastěji se jedná o poruchy spánku (63 %), v 69 % o poruchy usínání, 73 % vykazuje úzkostné a 42 % destruktivní chování. Velice častá je hyperaktivita (76 %), umíněnost až tvrdohlavost (47 %) a agrese (42 %).12 Viscerální postižení MPS II lze zmírnit nebo při časné diagnóze mu zcela předejít enzymovou substituční terapií. Postižení CNS zatím touto terapií ovlivnit nelze, zkouší se intratékální podávání enzymu.13
Mukopolysacharidóza typ III, Sanfilippo syndrom
Jedná se o skupinu čtyř fenotypově stejných, ale geneticky odlišných onemocnění, která vedou k poruše degradace heparansulfátu. Genetická podstata mukopolysacharidózy typ HIC byla objasněna na našem pracovišti teprve nedávno.14 Fyzické orgánové projevy MPS III jsou mnohem méně nápadné než u ostatních mukopolysacharidóz, naopak poruchy chování jsou prakticky konstantní (69-97 %).12 Onemocnění nastupuje po bezpříznakovém kojeneckém období nejčastěji mezi druhým a třetím rokem života opožděním vývoje a poruchami chování. Opoždění vývoje řeči většinou předchází motorickému opoždění. Typickými poruchami chování jsou neklid a hyperaktivita, agresivita, afektivní záchvaty s neobvyklým patologickým střídáním emocí (smích/křik/pláč) a orálním komplexem. Rodinu dítěte pak vyčerpává absence cirkadiánní rytmicity s převráceným spánkovým režimem s prevalencí 78-96 %. Děti mají problémy s usínáním, časně se budí, probouzejí se a bdí v noci, žvýkají ložní prádlo, pláčou, zpívají či se v noci smějí.12 Existují i vzácnější adultní formy MPS III, diagnostikované nejčastěji do třetí dekády, jeden pacient byl diagnostikován dokonce v 66 letech.15 Léčba pro MPS III t. č. není dostupná, zkouší se chaperonová terapie, substrát redukující terapie a intratékální podávání deficitního enzymu.
Onemocnění se u nyní 26leté dívky projevilo po období normálního vývoje v předškolním věku dyspraxií, impulzivitou, poruchou soustředění a hyperaktivitou. Tyto příznaky vedly k diagnóze poruchy pozornosti (Attention Deficit Disorder, ADD) a započata byla léčba methylfenidátem. Došlo ke zvýšení impulzivity nepozornosti, epizodickým emočním výbuchům, opozičnímu chování, sociální nezralosti a rozvoji jobických stavů. Při komplexním psychologickém vyšetření byla popsána jako hovorná, kooperativní a impulzivní, dosahovala 92 bodů IQ. Její diagnóza ADD byla rozšířena o pervazivní vývojou poruchu a poruchu opozičního vzdoru. V rozmezí 9. a 14. roku věku její IQ pokleslo a ustálilo se na 62 bodech až do 21 let. Její řeč byla plynulá, ale obsahovala řadu gramatických a syntaktických atypií. Během dalších dvou let zcela ztratila schopnost konverzovat a zvládat běžné denní aktivity (oblékání, čištění zubů a toaleta).16
Niemannova-Pickova nemoc typ C
Niemannova-Pickova nemoc typ C (NPC) je autosomálně recesivně děděné onemocnění lipidového metabolismu způsobené mutacemi v genu NPC1 (cca 95 % případů), NPC2 (cca 5 % případů). Incidence onemocnění je minimálně 1 : 120 000. Příčinou onemocnění je porucha esterifikace cholesterolu, intracelulárního lipidového transportu a následná akumulace sfingolipidů a řady jiných lipidů. Klinicky se jedná o multisystémové onemocnění sleziny, jater a CNS. K vysokému podezření na NPC vede nejčastěji kombinace splenomegalie a některého z příznaků postižení CNS: vertikální supranukleární obrna pohledu, nemotornost až neohrabanost, psychóza podobná schizofrenii, psychotické symptomy, kognitivní deficit, ataxie s dystonií, dysartrií či dysfagií.17 U pacientů s NPC s adultní psychiatrickou manifestací je často definitivní diagnóza stanovena opožděně, někdy až po 10 letech.18 Dle věku při prvním příznaku se rozlišuje časně infantilní forma (2 měsíce až 2 roky), pozdně infantilní forma (2-6 let), juvenilní (6-12 let), adolescentní/adultní (> 12 let). Věk nástupu onemocnění se může i mezi sourozenci výrazně lišit. Kauzální léčba NPC neexistuje, užívá se substrát redukující terapie preparátem miglustat, který stabilizuje nebo zpomaluje progresi neurologických příznaků onemocnění.17 Konstantním příznakem u všech pacientů je kognitivní deficit. U adultních forem NPC dochází na začátku k poruše exekutivních funkcí, následovaného poruchami paměti a globálním zpomalením kognitivního výkonu. U dospělých pacientů mohou být iniciální změny diskrétní. Poruchy exekutivních funkcí zahrnují časnou desinhibici, perservaci, chabý úsudek, ztrátu náhledu, ztrátu schopnosti abstrakce, dále bývá porušena pozornost. Prvním příznakem je často desinhibice. U dětí se pokles kognitivních funkcí projeví jako regres nebo opoždění psychomotorického vývoje. U dětí může být součástí obrazu NPC ještě před manifestací ostatních příznaků porucha pozornosti s hyperaktivitou (Attention-Deficit Hyperactivity Disorder, ADHD), rušivé či agresivní chování.19 Časté jsou poruchy paměti, zahrnující porušenou impregnaci vzpomínek a dezorientaci.20 Psychotické symptomy se u pacientů s NPC typicky manifestují v časné dospělosti či adolescenci, onemocnění může být rezistentní na léčbu antipsychotiky a pacienti bývají více sensitivní k vedlejším nežádoucím účinkům užívaných neuroleptik (zejména dystonie), jejichž léčba může být doprovázena sekundárními příznaky (např. vizuální halucinace, katatonie). U dětí a adolescentů s NPC byly popsány psychotické příznaky a mohou být přítomny současně s pervazivní vývojovou poruchou. Suspekci na toto onemocnění lze u 75 % pacientů vyslovit nalezením zvýšené aktivity plazmatické chitotriosidázy či hladiny oxysterolů, nově dostupná je analýza lysosphingomyelinu v suché krevní kapce tandemovou hmotnostní spektrometrií. Diagnózu potvrdíme molekulárně-genetickým vyšetřením. Vyšetření filipinovým barvením ve fibroblastech či hledáním pěnových buněk v punktátu kostní dřeně se v současné době již nepovažuje za nezbytné.
Tři roky depresivní a hypersomní pacientka byla hospitalizována ve věku 49 let pro emoční labilitu, logorrhoe, bludy a nadměrnou ostražitost. Byla léčena thiothixinem, imipraminem a lithiem. Ve věku 50 let se zhoršila stabilita chůze, což bylo diagnostikováno jako tardivní parkinsonismus a léčeno levodopou, na které se stav zlepšil. Novým příznakem byla dysfagie s nutností zavedení gastrostomie. Objevily se auditivní halucinace, paranoidní prožívání a nadměrná religiozita, byla léčena thioridazinem a amytriptylinem. Od 55 let se stav dále zhoršoval poruchou sakkadických očních pohybů, ataktickou dysartrií, rigiditou, bradykinezí, křečemi, demencí, mutismem. Diagnóza NPC byla stanovena až ve věku 61 let na základě biopsie kostní dřeně se specifickým nálezem pěnových buněk a analýzou kultivovaných fibroblastů s pozitivním nálezem ve specifickém filipinovém barvení. Zemřela ve věku 62 let.21
GM2 gangliosidóza, Tayova-Sachsova nemoc
Jedná o lysosomální onemocnění, které se vyznačuje střádáním nedegradovaných gangliosidů pocházejících z buněčných membrán neuronů. Klasická infantilní forma se vyznačuje zástavou vývoje, hypotonií a úlekovými reakcemi a úmrtím do 5 let života. Neurologická prezentace pozdní (juvenilní/adultní) formy GM2 gangliosidózy je variabilní, vyskytují se poruchy chůze, dysartrie, tremor. Úroveň kognice může být normální nebo blízká normě vykazující pouze mírné deficity exekutivních funkcí, psychomotorického tempa a verbální paměti. Zobrazení CNS prokáže u většiny pacientů atrofii mozečku. Diagnóza je stanovena vyšetřením enzymové aktivity v plazmě, leukocytech nebo kultivovaných fibroblastech. Onemocnění není léčitelné.22 Psychiatrické příznaky se vyskytují u poloviny pacientů s pozdní formou. Prvním příznakem může být katatonie, bipolární porucha, nejčastěji se však jedná o psychózu podobnou schizofrenii na organickém podkladě (30-50 %). Častá je kombinace PM s neuropath. Komplikovaná je terapie s nedostatečným efektem lithia a vysokou citlivostí pacientů na extrapyramidové vedlejší účinky antipsychotik.6
Metachromatická leukodystrofie
Metachromatická leukodystrofie je další zástupce skupiny lysosomálních střádavých poruch. Charakteristický je obraz na MRI s typickými splývajícími změnami bílé hmoty šetřící subkortikální U vlákna. Leukodystrofie mívá frontotemporální převahu a minimální postižení šedé hmoty. Klasická pozdně infantilní forma se projeví regresem v batolecím věku, svalovou atrofii, rigiditou, ztrátou zraku, křečemi a úmrtím do pěti let věku.23 Adultní forma se manifestuje dvěma fenotypy - predominantně motorickým deficitem (mozečkovo-pyramidálním) nebo psychiatrickým. Klasický nález leukodystrofie na MRI CNS nebývá u této formy onemocnění přítomný. Až polovina pacientů mezi 10 a 30 lety má psychotické symptomy zahrnující sluchové halucinace, bludy, formální poruchy myšlení a katatonii. Ty často předcházejí křečím, choree nebo dystonii.24 Pozitivita metachromatických substancí v moči či nález zvýšené sulfatidurie ujistí naše podezření na biochemické úrovni, diagnóza je následně potvrzena enzymatickým vyšetřením arylsulfatázy A v leukocytech nebo fibroblastech a molekulárně-genetickým vyšetřením.
X-vázaná adrenoleukodystrofie
X-vázaná adrenoleukodystrofie je peroxisomální onemocnění zapříčiněné mutacemi v genu ABCD1 pro peroxisomální membránový přenašeč. I když základním patofyziologickým podkladem je nejspíše akumulace mastných kyselin s velmi dlouhým řetězcem (Very Long-Chain Fatty Acids, VLCFA), výsledný fenotyp určuje celá řada sekundárních procesů (mj. oxidační stres, nedostatek energie v axonech, zánětlivá složka demyelinizace, porucha integrity hematoencefalické bariéry). Dětská a adolescentní forma se vyznačují progresivní neuro degenerací do vegetativního stadia, liší se pouze nástupem onemocnění (cerebrální forma). Onemocnění se může projevit i jako adrenomyelopatie s progresivní neuropatií, paraparézou a depresí, kdy do postižení CNS dospěje asi 40 % pacientů. X-vázaná adrenoleukodystrofie má charakteristický obraz na MRI se symetrickými hyperintenzitami v posteriorní části corpus callosum, které se šíří do parietookcipitálních oblastí v T2 vážených sekvencích.25 Adultní forma se typicky manifestuje psychiatrickými poruchami, nejčastěji změnami chování. Mánie a schizoafektivní porucha jsou nejčastějšími poruchami, ale později se vyskytují i čistě schizofreniformní poruchy. Někteří pacienti mohou mít změny nálady v důsledku adrenální insuficience, které lze vyléčit odpovídají hormonální suplementací. U žen se příznaky vyskytují v cca 65 %, zejména pod obrazem adrenomyelopatie a chronické bolesti připomínající fibromyalgie.24 Stanovení hladiny velmi dlouhých mastných kyselin (zejména kyseliny hexakosanové C26) v séru je základem pro vyslovení suspekce, diagnóza se poté potvrdí na molekulárně-genetické úrovni.
Wilsonova choroba
Wilsonova choroba, dříve označována jako hepatolentikulární degenerace, je autosomálně recesivně děděné onemocnění s incidencí 1 : 30 000.26 Důvodem jsou mutace v genu ATP7B kódujícím přenašeč mědi. Organismus pak nedostatečně vylučuje měď žlučí a ta se ukládá v játrech, centrálním nervovém systému, ledvinách a v kostech. Onemocnění se nejčastěji manifestuje u preadolescentních dětí, adolescentů a mladých dospělých, opět však byly popsány vzácné kazuistiky s pozdní manifestací v sedmé dekádě života. Přibližně jedna třetina pacientů se projeví jaterní nemocí, jedna třetina neurologickými příznaky (tremor, dysartrie, dystonie, parkinsonský syndrom, spasticita) a jedna třetina psychiatrickou symptomatologií (viz dále). Časté jsou rovněž oftalmologické příznaky (Kayser-Fleischerův prstenec, slunečnicová katarakta), renální (urolitiáza, hematurie), kostní (chondrokalcinóza, osteoartritida) a hematologické (akutní hemolytická anémie, trombocytopenie). Charakteristickým obrazem na MRI CNS jsou hyperintenzní ložiska ve striatu, globus pallidus, pontu a mezencefalu, méně často i v talamech a mozečku v T2 vážených obrazech.26
Psychiatrické příznaky se ve skupině 195 pacientů vyskytly v 51 %, neurologickým příznakům předcházely až ve 20 % případů.27 Hlavními symptomy byly poruchy chování (iritabilita, agresivita, výrazné změny osobnosti, disinhibice) nebo depresivní syndrom. Psychózy jsou poměrně vzácné, pouze v 1 % případů.28
Na nasazené chelatační terapii dochází ke stabilizaci nebo zlepšení příznaků, psychiatrické problémy se však i na terapii mohou dále horšit.29 Farmakologická léčba Wilsonovy nemoci je spojována s celou řadou komplikací a omezení. Neuroleptika mohou zvyšovat riziko akinezy a rigidity při postižení bazálních ganglií. Použití atypických neuroleptik (olanzapin, risperidon, quetiapin, klozapin) se považuje z tohoto pohledu za poměrně bezpečné, i když při použití těchto léků zase hrozí zvýšené riziko agranulocytózy při hypersplenismu nebo léčbě penicilaminem. Valproát a karbamazepin mohou být kontraindikovány při jaterním postižení, lithium pak v přítomnosti renální tubulární acidózy.30
12letý chlapec byl přijatý pro distenzi břicha spojenou s asi měsíc trvající bolestí. Rozvinula se rovněž podrážděnost, bludná vztahovačnost i strukturované paranoidně-persekuční bludy se značným funkčním dopadem (odmítal docházet do školy). Základní laboratorní vyšetření bylo v pořádku, ale ultrasonografické vyšetření břicha prokázalo změnyparenchymu, portální hypertenzi a ascites. Diagnóza Wilsonovy nemoci byla stanovena na základě nízké sérové hladiny ceruloplasminu a volné mědi a zvýšeného odpadu mědi v moči. Vyšetření štěrbinovou lampou prokázalo Kayser-Fleischerův prstenec. Po dvou týdnech se plně rozvinula akutní psychotická ataka s paranoidně-persekučními bludy, bázlivostí, ostražitostí a poruchou spánku. Po počátečním zhoršení na chelatační léčbě penicilaminem došlo po redukci dávky a nasazení risperidonu k ústupu psychiatrických příznaků.31
MITOCHONDRIÁLNÍ ONEMOCNĚNÍ
Mitochondriální onemocnění (MO) energetického metabolismu představují klinicky a biochemicky heterogenní skupinu způsobenou mutacemi ve více než 260 genech nukleární nebo mitochondriální DNA. Variabilita klinických příznaků je široká, nejčastěji je přítomna kombinace postižení orgánů s vysokými energetickými nároky jako mozek, srdce, sval a smysly (obr. 2). Onemocnění se opět mohou projevit v nejtěžší podobě v novorozeneckém věku, ale i v oligosymptomatických formách manifestujících se u dospělých jedinců.32 Jedná se o postižení chronické, může však docházet k akutním krizím s iktu podobnou příhodou, laktátovou acidózou, případně hyperamonémií,33,34 které se mohou projevit náhlou neurologickou nebo i psychiatrickou symptomatikou. Stanovení diagnózy MO může být komplikováno jejich nespecifickou manifestací. Spouštěčem manifestace může být psychický nebo fyzický stres, onemocnění se může projevit svalovou slabostí nebo intolerancí fyzické námahy, chronickou únavou, bolestmi hlavy, zad, nadměrnou spavostí, nevýkonností a závratí.35 Jedná se také často o mírné a nekompletní formy MO - např. pouze 14 % českých pacientů se syndromem MELAS (Mitochondriální myopatie, Encefalopatie, Laktátová Acidóza a Stroke-like Episodes neboli Iktu podobné příhody) prodělalo nejtěžší komplikaci onemocnění - iktu podobné příhody.33 Dokonce i normální hladina laktátu v krvi MO nevylučuje, zvláště v případě mírných a pozdně se manifestujících forem.36 I když MO nedokážeme vyléčit, v některých případech má správná diagnóza pro pacienta zcela zásadní význam. Příkladem může být suplementace argininu či citrulinu v případě MELAS syndromu, která prokazatelně snižuje incidenci iktu podobných příhod a jeho intravenózní podání ve vysokých dávkách v případě akutní epizody zmírňuje její klinický dopad. Zásadní význam má i genetické poradenství v rodině.
Celoživotní prevalence psychiatrických poruch ve skupině mitochondriálních onemocnění dosahuje 47 až 69 %.35,37,38 Frekvence psychiatrických poruch je tak vyšší nejenom ve srovnání s běžnou populací (20 %),37 ale i se skupinami klinicky podobně postižených pacientů (30 %).38 Nejčastější psychiatrickou morbiditou ve skupině MO je deprese, např. 28 % ve skupině 71 pacientů se syndromem MELAS;39 54 % ve skupině 36 pacientů s MO.35 Deprese se může projevit již u dětí s MO; byla diagnostikována u 5 z 35 dětí s MO.40 Rekurentní deprese byla dokonce prvním příznakem jednoho z našich pacientů s MELAS syndromem.
Chlapec byl od batolecího věku méně výkonný, později nestačil vrstevníkům, přestal růst, nápadná byla výrazná únava a svalová slabost. V osmi letech se přidaly bolesti břicha s opakovaným zvracením. Klinicky byl přítomný centrální hypotonický syndrom, snížená svalová síla, svalová hypotrofie. Laboratorně byla přítomna myopatie. Bylo vysloveno podezření na mitochondriální onemocnění, prokázán byl syndrom MELAS s nálezem prevalentní mutace 3243A>G v mitochondriální DNA. Nemoc se komplikovala epilepsií a migrenózními bolestmi hlavy od 11 let. Ve 13 letech chlapec poprvé prodělal iktu podobnou příhodu, která se projevila těžkou čtyři dny trvající bolestí hlavy, zvracením, poruchou vizu, únavou s excesivní spavostí, hemianopsií, nystagmem, afázií, dysartrií a dysfázií. Do 17. roku života se opakovaly další tři iktu podobné příhody, jejichž důsledkem se rozvinula periferní a centrální sluchová porucha a sekundární epilepsie. Od 16 let se rozvinula externí oftalmoplegie a ptóza víček, klinicky i EMG vyšetřením potvrzena polyneuropatie s hyporeflexií, myopatií, rozvinutá je mozečková symptomatologie. Příznaky byly doprovázeny těžkou depresí se ztrátou zájmů a komunikačními obtížemi. Intelekt je v pásmu lehkého opoždění.41
V literatuře najdeme i několik kazuistik pacientů s MELAS syndromem (nebo jinými onemocněními s mutacemi v mitochondriální DNA), kteří se manifestovali stavy připomínajícími psychózu charakterizovanou bludy, halucinacemi, zmateností, dezorganizovanou řečí, přehnanou nebo oploštělou emotivitou, bizarním chováním či psychomotorickou excitací nebo naopak stuporem.42 Opět uvádíme kazuistiku z naší praxe.
MELAS syndrom se u 16leté dívky manifestoval migrenózními bolestmi hlavy, zvracením a epilepsií. Prodělala několik iktu podobných příhod, rozvinul se paleocerebelární syndrom, intermitentní nystagmus, porucha sluchu a rozmazané vidění. Její kognice byla deteriorovaná. Byla přijata na psychiatrické lůžko ve věku 29 let pro rychle progredující dezorientaci s halucinatorním syndromem. Výrazné bylo diurnální kolísání intenzity příznaků. Nebyla schopna produktivní komunikace, odpovídala mimo relaci dotazů. Nálada byla výrazně labilní, po většinu času byla dysforická a často plakala, mnohdy byl přítomen neadekvátní smích. Chování bylo infantilní, odmítala otevírat oči. Tato epizoda postupně odezněla, pacientka byla diagnostikována s organickou schizofrenii podobnou poruchou. Přes dlouhodobou léčbu pregabalinem, olanzapinem a citalopramem nebylo zabráněno dalším hospitalizacím na psychiatrickém oddělení.41
ZÁVĚR - SHRNUTÍ PRO PRAXI

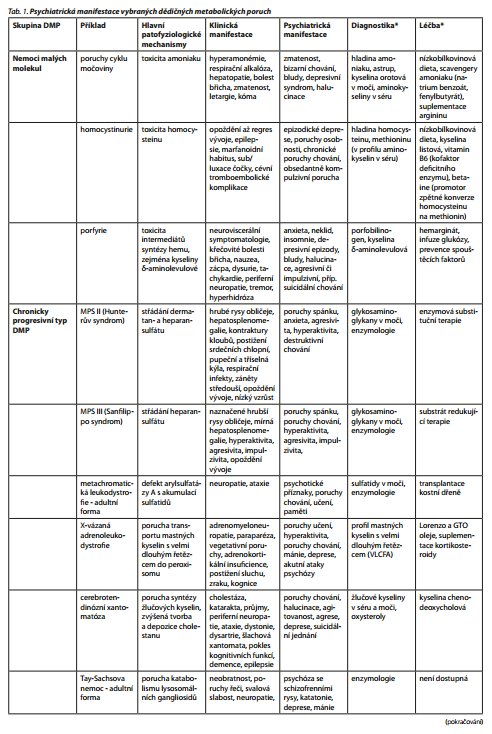
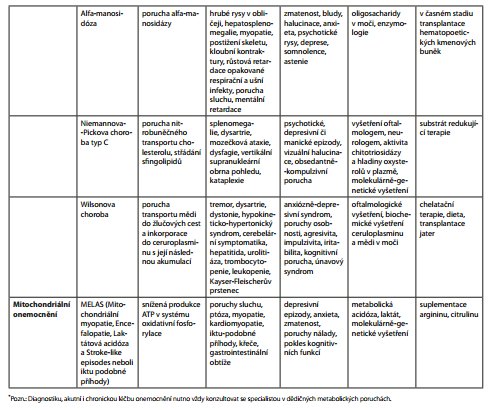
Diagnostika DMP je náročná (tab. 1). O to více může být komplikována v dospělém věku, kdy se často jedná o oligosymptomatické formy, u kterých může být PM jediným příznakem. Diagnostikovat DMP pak nemusí být jednoduché ani pro specialistu se zkušeností v daném oboru. K tomu se jedná o onemocnění vzácná, která se v převaze "neorganických" pacientů snadno ztratí. Správná diagnóza se zavedením adekvátní terapie však může zcela zásadně změnit osud pacienta a často i jeho rodiny. Lze vysledovat alespoň několik rysů, které by měly lékaře navést k širší diagnostice organické poruchy (tab. 2). Např. u organických poruch se častěji vyskytují vizuální halucinace a zmatenost, zatímco u schizofrenie jsou mnohem častější sluchové halucinace, nebo alespoň jsou nad zrakovými v převaze.43 Zvláště u akutně intoxikačního typu DMP je nástup psychiatrických příznaků náhlý častokrát ve spojitosti se zátěží organismu (infekce, operace, porod) nebo změnou diety. Psychiatrické příznaky u DMP většinou začínají v nižším věku, často jsou doprovázeny poklesem kognitivních funkcí.44 Na DMP nutno myslet zvláště u multisystémových projevů (např. neobjasněná organomegalie - lysosomální střádavá onemocnění; postižení srdce a svalů, ptóza - porucha mitochondriálního energetického metabolismu). Dále jsou to atypické projevy, např. katatonie v dětství nebo adolescenci (např. Wilsonova choroba, NPC, GM2 gangliosidóza) nebo supranukleární obrna vertikálního pohledu (NPC). Pomyslnou červenou vlaječkou může být i fakt, že onemocnění reaguje špatně nebo vůbec na běžně podávanou farmakologickou terapii a často závažnější bývají extrapyramidové nežádoucí účinky antipsychotik. Při podezření na DMP lze pacienta telefonicky konzultovat s lékařem Ústavu dědičných metabolických poruch 1. lékařské fakulty Univerzity Karlovy a Všeobecné nemocnice v Praze na telefonním čísle 224 967 230, e-mailové adrese tomas.honzik@vfn.cz, udmp@vfn.cz, nebo přímo objednat ambulantní vyšetření na telefonním čísle 224 967 669. U dětí možno domluvit vyšetření ambulantní nebo během hospitalizace s lékařem metabolické jednotky Kliniky dětského a dorostového lékařství ( tomas.honzik@vfn.cz, tel. 224 967 792 či martin.magner@vfn.cz, 224 967 791).
LITERATURA
- 1. Honzík T, Zeman J. Dědičné metabolické poruchy v dětském věku. Praha: Institut postgraduálního vzdělávání ve zdravotnictví; 2013: 1-96.
- 2. Fletcher PC, Frith CD. Perceiving is believing: a Bayesian approach to explaining the positive symptoms of schizophrenia. Nat Rev Neurosci 2009; 10 (1): 48-58.
- 3. Storkanova G, Vlaskova H, Chuzhanova N et. al. Ornithine carbamoyltransferase deficiency: molecular characterization of 29 families. Clin Genet 2013; 84 (6): 552-559.
- 4. Arn PH, Hauser ER, Thomas GH et al. Hyperammonemia in women with a mutation at the ornithine carbamoyltransferase locus. A cause of postpartum coma. N Engl J Med 1990; 322 (23): 1652-1655.
- 5. Vítek L. Diagnostika porfyrické nemoci - Část 2. FONS 2008; 18 (2): 23-28.
- 6. Walterfang M, Bonnot O, Mocellin R, Velakoulis D. The neuropsychiatry of inborn errors of metabolism. J Inherit Metab Dis 2013; 36 (4): 687-702.
- 7. Kumar B. Acute intermittent porphyria presenting solely with psychosis: a case report and discussion. Psychosomatics 2012; 53 (5): 494-498.
- 8. Magner M, Krupková L, Honzík T et al. Vascular presentation of cystathionine beta-synthase deficiency in adulthood. J Inherit Metab Dis 2011; 34 (1): 33-37.
- 9. Abbott MH, Folstein SE, Abbey H, Pyeritz RE. Psychiatric manifestations of homocystinuria due to cystathionine beta-synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitamin B6-responsiveness. Am J Med Genet 1987; 26 (4): 959-969.
- 10. Huemer M, Mulder-Bleile R, Burda P et al. Clinical pattern, mutations and in vitro residual activity in 33 patients with severe 5, 10 methylenetetrahydrofolate reductase (MTHFR) deficiency. J Inherit Metab Dis. In press 2015.
- 11. Magner M, Hrubá E, Poupětová H et al. Klinická manifestace Hunterovy nemoci u 22 českých pacientů. Čes-slov Pediat 2014; 69 (SI): 56.
- 12. Cross EM, Hare DJ. Behavioural phenotypes of the mucopolysaccharide disorders: a systematic literature review of cognitive, motor, social, linguistic and behavioural presentation in the MPS disorders. J Inherit Metab Dis 2013; 36 (2): 189-200.
- 13. Kulhánek J, Malinová V, Honzík T, Magner M. Enzymová substituční terapie u lysosomálních onemocnění. Čes-slov Pediat 2015; 70 (4): 224-231.
- 14. Hřebíček M, Mrázová L, Seyrantepe V et al. Mutations in TMEM76* cause mucopolysaccharidosis HIC (Sanfilippo C syndrome). Am J Hum Genet 2006; 79 (5) : 807-819.
- 15. Valstar MJ, Bruggenwirth HT, Olmer R et al. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis 2010; 33 (6): 759-767.
- 16. Brady J, Trehan A, Lan dis D, Toro C. Mucopolysaccharidosis type IIIB (MPS IIIB) masquerading as a behavioural disorder. BMJ Case Rep. [online] May 2013 [cit. 2015-09-29]. Dostupné na Internetu: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3669838/.
- 17. Mengel E, Klünemann HH, Lourenco CM et al. Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J Rare Dis 2013; 8: 166.
- 18. Klünemann HH, Santosh PJ, Sedel F. Treatable metabolic psychoses that go undetected: what Niemann-Pick type C can teach us. Int J Psychiatry Clin Pract2012; 16(3): 162-169.
- 19. Albrecht J, Anders M. Psychické projevy Niemannovy-Pickovy choroby typu G Čes a slov Psychiat 2011; 107 (6): 346-353.
- 20. Mattson MP, Shea TB: Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 2003; 26 (3): 137-146.
- 21. Josephs KA, Van Gerpen MW, Van Gerpen JA. Adult onset Niemann-Pick disease type C presenting with psychosis. J Neurol Neurosurg Psychiatry 2003; 74 (4): 528-529.
- 22. Patterson MC. Gangliosidoses. Handb Clin Neurol 2013; 113: 1707-1708.
- 23. van Rappard DF, Boelens JJ, Wolf NI. Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab 2015; 29 (2): 261-273.
- 24. Rosebush PI, Mazurek ME Catatonia and its treatment. Schizophr Bull 2010; 36 (2): 239-242.
- 25. Berger J, Gartner J. X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta2006; 1763 (12): 1721-1732.
- 26. Dušek P, Brůha R, Burgetová A, Záhoráková D, Růžička E. Wilsonova nemoc. Cesk Slov Neurol N 2013; 76/109 (5): 539-549.
- 27. Walshe JM, Yealland M. Wilson's disease: the problem of delayed diagnosis. J Neurol Neurosurg Psychiatry 1992; 55: 692-696.
- 28. Denning TR, Berrios GE. Potential confusion of neuroleptic malignant syndrome and Wilson's disease. Lancet 1989; 2: 43.
- 29. Smith RJ, Bryant RG: Metal substitutions incarbonic anhydrase: a halide ion probe study. Biochem Biophys Res Commun 1975; 66 (4): 1281-1286.
- 30. Varghese ST, Narayanan D, Dinesh D: Mania in a patient with Wilson's disease awaiting liver transplant. J Neuropsychiatry Clin Neurosci 2008; 20 (4): 501-502.
- 31. Grover S, Sarkar S, Jhanda S, Chawla Y. Psychosis in an adolescent with Wilson's disease: A case report and review of the literature. Indian J Psychiatry 2014; 56 (4): 395-398.
- 32. Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic communication. Biosci Rep 2007; 27 (1-3): 39-51.
- 33. Dvořáková V, Tesařová M, Honzík T. MELAS syndrom. In: Vzácná onemocnění II. Olomouc: Solen; 2014: 44-56.
- 34. Magner M, Dvořáková V, Tesařová M et al. TMEM70 deficiency: long-term outcome of 48 patients. J Inherit Metab Dis 2015; 38 (3): 417-426.
- 35. Fattal O, Link J, Quinn K, Cohen BH, Franco K. Psychiatric comorbidity in 36 adults with mitochondrial cytopathies. CNS Spectr 2007; 12 (6): 429-438.
- 36. Magner M, Szentiványi K, Švandová I et al. Elevated CSF-lactate is a reliable marker of mitochondrial disorders in children even after brief seizures. Eur J Paediatr Neurol 2011; 15 (2): 101-108.
- 37. Mancuso M, Orsucci D, Ienco EC et al. Psychiatric involvement in adult patients with mitochondrial disease. Neurol Sci 2013; 34 (1): 71-74.
- 38. Inczedy-Farkas G, Remenyi V, Gal A et al. Psychiatric symptoms of patients with primary mitochondrial DNA disorders. Behav Brain Funct; 2012; 8: 9.
- 39. de Laat P, Koene S, van den Heuvel LPWJ et al. Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243A>G mutation. J Inherit Metab Dis 2012; 35 (6): 1059-1069.
- 40. Koene S, Kozicz TL, Rodenburg RJ et al. Major depression in adolescent children consecutively diagnosed with mitochondrial disorder. J Affect Disord 2009; 114 (1-3): 327-332.
- 41. Magner M, Honzík T, Tesařová M et al. Psychiatric disturbances in five patients with MELAS syndrome. Psychiatr Pol 2014; 48 (5): 1035-1045.
- 42. Kaufman KR, Zuber N, Rueda-Lara MA, Tobia A. MELAS with recurrent complex partial seizures, nonconvulsive status epilepticus, psychosis, and behavioral disturbances: case analysis with literature review. Epilepsy Behav 2010; 18 (4) : 494-497.
- 43. Bonnot O, Klünemann HH, Sedel F et al. Diagnostic and treatment implications of psychosis secondary to treatable metabolic disorders in adults: a systematic review. Orphanet J Rare Dis [online] Apr 2014; 9: 65. [cit. 2015-09-29]. Dostupné na Internetu: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4043981/
- 44. Remschmidt H, Theisen F. Early-onset schizophrenia. Neuropsychobiology 2012; 66 (1): 63-69.